
PadeNA: A PARALLEL DE NOVO ASSEMBLER
Gaurav Thareja, Vivek Kumar
Aditi Technologies, Manyata Tech Park, Bangalore, India
Mike Zyskowski, Simon Mercer, Bob Davidson
Microsoft Research, One Microsoft Way, Redmond, WA 98052, U.S.A.
Keywords: DNA Sequence Assembly, Genomics, de Bruijn Graph, Scaffold Generation.
Abstract: Recent technological advances in DNA sequencing technology are resulting in ever-larger quantities of
sequence information being made available to an increasingly broad segment of the scientific and clinical
community. This is in turn driving the need for standard, rapid and easy to use tools for genomic
reconstruction and analysis. As a step towards addressing this challenge, we present PadeNA (Parallel de
Novo Assembler), a parallelized DNA sequence assembler with a graphical user interface. PadeNA is
designed using interface-driven architecture to facilitate code reusability and extensibility, and is provided
as part of the open source Microsoft Biology Foundation. Installers and documentation are available at
http://research.microsoft.com/bio/.
1 INTRODUCTION
Many attempts have been made to address the DNA
sequence assembly problem and all give way to
heuristic methods at some stage. Traditionally,
Sanger sequencing projects have relied on a heuristic
assembly method known as overlap-layout-
consensus, in which overlaps between reads are used
to guide the assembly. This graph-based model has
inspired the development of applications such as the
TIGR (Sutton, 1995), Celera (Myers 2000), Phrap
(Green, 1996), CAP3 (Huang, 1999), Atlas (Havlak,
2003) and ARACHNE (Batzoglou, 2002)
assemblers.
The latest generation of DNA sequencing
technologies is capable of producing far greater
volumes of data, but these tend to be in the form of
short sequence reads. With short reads eliminating
the reliability of read overlaps, the pioneering work
of Pevzner et al. (Pevzner, 2001) on de Bruijn
graphs now forms the basis of many of the current
generation of short-read assemblers. Velvet
(Zerbino, 2008), ALLPATHS (Butler 2008), Euler
SR (Chaisson, 2008) and ABySS (Simpson, 2009)
all have de Bruijn graphs at the heart of their
algorithms.
Many of the currently available short read de novo
assemblers are single-threaded applications
accessible through a command line interface and
designed to run on a single processor or distributed
memory architectures. While sufficient for the
current requirements of genomics, the increasing
availability of cheap DNA sequence is already
revolutionizing the many branches of genomics
research and finding increasingly broad application
in healthcare and non-traditional fields from
environmental studies to law enforcement. This
radically broadened user base will demand tools that
are compatible with the latest sequencing
technologies, are adapted to their specific needs and
are responsive and intuitive. This in turn requires a
code base that can leverage the capabilities of
computer hardware with shared memory
architectures and facilitate rapid application
development and intuitive user interface design.
Many of these needs can be met within a modular
framework of reusable bioinformatics componentry
open to community contribution and freely available
for both commercial and academic developers. Such
a framework would be able to evolve along with the
technologies it supported, extending as needed to
accommodate new experimental techniques and
computer architectures while reducing the level of
196
Thareja G., Kumar V., Zyskowski M., Mercer S. and Davidson B..
PadeNA: A PARALLEL DE NOVO ASSEMBLER.
DOI: 10.5220/0003164301960203
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2011), pages 196-203
ISBN: 978-989-8425-36-2
Copyright
c
2011 SCITEPRESS (Science and Technology Publications, Lda.)

effort needed to support an increasingly diverse user
community.
2 IMPLEMENTATION
PadeNA (the Parallel de Novo Assembler) has been
implemented with the principles of code
modularization and reusability in mind, and focuses
on the concept of parallelization in shared memory
architecture. This is the first application of its kind
developed for Windows-based users. The major
improvement in PadeNA is interface driven design
(Pattison, 1999) which promotes reusability and
extensibility of code without affecting its data
handling capabilities. In effect, PadeNA is a
sequence of more basic functions, and developers
can easily customize the default algorithm by
inserting or substituting their own custom classes to
meet the needs of their users. In order to
demonstrate the functionality of PadeNA, we have
also developed a graphical user interface using
Windows Presentation Foundation, providing a
usable and intuitive interface to this and other
assembly algorithms.
PadeNA is built as a part of a .NET based open-
source bioinformatics library, the Microsoft Biology
Foundation (MBF). It uses .NET 4.0 constructs for
multi-core parallelization and performance scales
well on computers with two or more processors as
compared to single-core systems.
Microsoft .NET framework uses a method to
improve the runtime performance of computer
programs. This is known as just-in-time compilation
(JIT), also known as dynamic translation. JIT
compilers represent a hybrid approach, with
translation occurring continuously, as with
interpreters, but with caching of translated code to
minimize performance degradation. It also offers
other advantages over statically compiled code at
development time, such as handling of late-bound
data types and the ability to enforce security
guarantees.
Moreover, Native Image Generator, or simply
NGEN is the Ahead-of-time compilation service of
the .NET Framework. It allows a .NET assembly to
be pre-compiled instead of letting the Common
Language Runtime do a Just-in-time compilation at
runtime (Biswas, 2006).
Further, bioinformatics researchers working in
Unix/Linux environment can take advantage of
Mono 2.8 for extending PadeNA. Mono is an open
source, cross-platform, implementation of C# and
the CLR that is binary compatible with
Microsoft.NET (Mono, 2004).
2.1 Microsoft Biology Foundation
The Microsoft Biology Foundation is an open source
reusable .NET library and application programming
interface for bioinformatics research. Application
developers can use MBF to perform a wide range of
tasks; DNA, RNA and protein sequences can be
imported from files in a variety of standard data
formats, including FASTA, FASTQ, GenBank,
GFF, BED, SAM and BAM. Analysis of these
sequences can be performed using one of several
sequence alignment algorithms including Smith-
Waterman, Needleman-Wunsch, pairwise overlap
aligner, MUMmer and NUCmer (Kurtz, 2004).
These sequences can also be queried against various
databases using BLAST (Altschul, 1997) services,
hosted at different locations and accessible through a
web service interface. File formatters can be used to
write sequences in the desired supported output
format irrespective of the original input format.
Data files are sometimes large enough that hardware
limitations prevent a parser from loading the entire
data set into memory – this may occur when
handling one very large sequence, or a very large
file (or files) containing many smaller sequences.
MBF implements data virtualization by dividing the
data into blocks and providing the data block by
block to the parser as required by the application.
MBF represents sequence data and metadata with
format-independent Sequence and SequenceRange
objects. These objects efficiently store sequence data
in a variety of encoded formats and provide a
flexible and robust way to represent sequences in the
MBF environment.
MBF applications can be implemented in any .NET-
compatible language. Over 70 of these exist, suiting
many different programming styles and levels of
expertise; examples include C#, F#, Visual Basic
and Python.
2.2 Input Parameters
Kmer Length: The choice of kmer length is a
critical task. The search space depends upon kmer
length, with smaller kmer length increase the
number of vertices in the graph. Larger kmer length
reduces the number of ambiguous edges in the graph
but also significantly impact the true overlaps
between kmers. For optimal graph formation, kmer
length should not be less than half the length of the
PadeNA: A PARALLEL DE NOVO ASSEMBLER
197
longest input sequence and cannot be more than the
length of the shortest input sequence.
Dangle Threshold: Maximum length to traverse
from dead ends till point of ambiguity is reached.
This value is dependent on kmer length. (Default:
Kmer length + 1)
Redundant Path Length Threshold: Maximum
length to traverse from point of ambiguity till paths
converge in the graph. (Default: 3 * (Kmer length +
1))
Erosion Threshold: The parameter erodes bases at
the ends of blunt contigs with coverage less than the
specified threshold. (Default: Square root of median
of kmer coverage)
Contig Coverage Threshold:
The parameter
removes low coverage contigs. (Default: Square root
of median of kmer coverage)
Scaffold Redundancy:
The number of mate pair
connections required to connect a pair of contigs.
(Default: 2)
Depth:
This parameter defines the threshold while
performing depth first search on contig overlap
graph. (Default: 10)
3 ASSEMBLY ALGORITHM
Sequence assembly algorithms typically have two
major phases. In the first phase, contigs are extended
until either they cannot be unambiguously extended
further or they reach an end due to lack of read
coverage.
During the second phase, information from paired-
end reads is used to resolve ambiguities and order
and merge contigs to generate scaffolds. We have
used similar steps to those already available in
ABySS (Simpson, 2009), Euler SR (Chaisson,
2008), BAMBUS (Pop, 2004) and the Greedy Path
Merging Algorithm (Huson, 2002). However we
have parallelized many of them, as described later.
3.1 Building the de Bruijn Graph
The sequence reads are first loaded using the data-
virtualized parser of MBF and read sequences with
ambiguous characters are removed prior to
construction of the graph. The remaining sequences
are broken into kmers by defining a window of
length k and moving it along each sequence one base
at a time. The forward and reverse complementary
sequence of a kmer are considered equivalent. We
consider lexicographically larger kmer sequence as
sequence from positive strand.
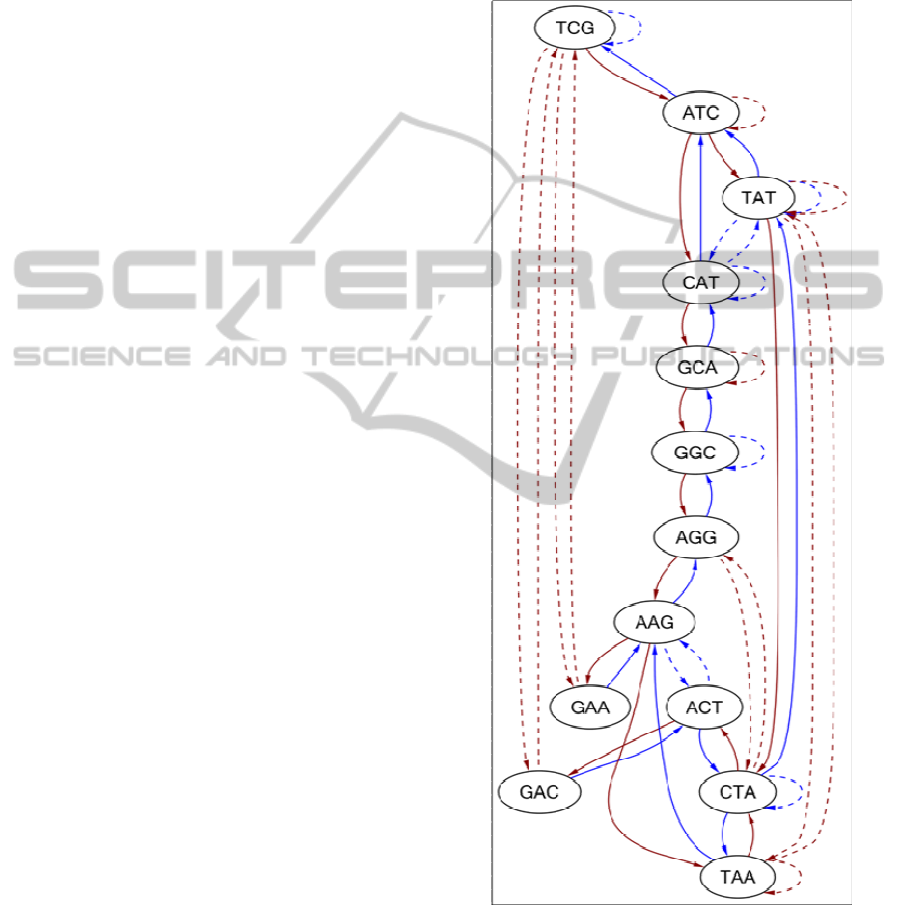
A de Bruijn Graph is a edit distance graph in which
nodes corresponds to objects, and two nodes are
connected if the edit distances between the objects
represented by those nodes is one. Each node in the
Figure 1: Bi-directed de Bruijn Graph for sequence
ATCGACTATAAGGCATCGAA using kmer length = 3.
Where, blue solid lines indicate forward edges, Brown
solid lines indicate backward edges, blue dotted lines
indicate forward edges for reverse complementary
sequence and brown dotted lines indicate backward edges
for reverse complementary sequence.
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
198

graph corresponds to a unique kmer present in some
input sequence or its reverse complement. A
directed edge connects two nodes labeled aα and αb,
where α is a string of length k-1. A bi-directed de
Bruijn Graph is a natural model for the assembly
problem because the two labels for nodes
corresponds two strands of DNA molecule.
3.2 Error Removal
This is the most critical step for assembly. The steps
involved here will remove sequencing errors in the
reads. This process also removes single nucleotide
polymorphisms.
3.2.1 Dangling Link Removal
Any kmer containing a sequencing error is likely to
be unique and will therefore have only a single
connection to a preceding kmer in the graph,
forming a short side branch. The graph is traced for
branches with dead ends and then these branches are
traced back to a point of ambiguity is reached. If the
count of the nodes traversed is less than threshold
value then nodes are removed from the graph. This
step is iteratively performed for bigger branches
with large number of nodes as removal of small
branches may create longer dangling links until the
threshold is reached. The effectiveness of the
dangling link removal step also depends on kmer
length (longer kmers may contain multiple sequence
errors, in turn creating longer dangling links).
3.2.2 Redundant Path Removal
Another common structure found in de Bruijn
graphs is often caused by sequencing errors or single
nucleotide polymorphisms in the middle of reads.
Sequence variation of this kind creates ambiguity in
the graph and impairs contig extension. The graph is
traversed to find all points of divergence, and at each
point of divergence the path is traced forward until a
user-defined threshold is reached. If the paths
converge and contain equal number of nodes, then
the path with lower coverage is removed from the
graph.
3.3 Contig Builder
In the final step of the first phase of sequence
assembly, the graph is traversed to find nodes with
ambiguous edges and these edges are removed,
breaking the graph into a number of sub graphs. The
DNA sequence of each sub-graph is reconstructed
from its constituent kmers to create the initial set of
contigs.
3.4 Scaffold Generation
The second phase takes into consideration mate-pair
information to determine an overall ordering of
contigs.
Libraries of DNA fragments used in sequencing are
frequently generated by experimental techniques that
guarantee all fragments fall within a defined size
range.
Mate-pairs are pairs of reads corresponding to the
DNA sequence at each end of the same fragment of
DNA and have a known strand and orientation
relative to each other. Where the mean and standard
deviation of fragment length is known for a library,
we can estimate the distance between reads in a
mate-pair and use this information to find an order
and orientation of all contigs – a process known as
scaffolding.
Mate pairs from multiple DNA libraries can also be
used in this step. Each library is considered for
distance estimation between contigs.
3.4.1 Aligning Reads to Contigs
Reads are aligned to contigs before attempting to
establish links between contigs. In PadeNA, we
create ungapped alignments by converting each read
to a list of kmers and matching these kmers to a list
of kmers similarly generated from the contig.
3.4.2 Establishing Mate-pair Links
between Contigs
Potential links between contigs are determined from
mate-pair information in the aligned reads. The
information is encoded in the read id, and PadeNA
supports several standard naming conventions.
3.4.3 Filtering Mate-pairs
Mate-pairs are initially filtered based on contig
orientation. The orientation of pair of contigs is the
orientation supported by the largest number of mate-
pairs that connect them. A mate-pair connection is
confirmed between a pair of contigs only if the
number of mate-pairs supporting a particular
orientation is greater than or equal to a threshold
value. This removes spurious links between contigs
and helps in the correct estimation of distance
between contigs. (Pop, 2004)
PadeNA: A PARALLEL DE NOVO ASSEMBLER
199
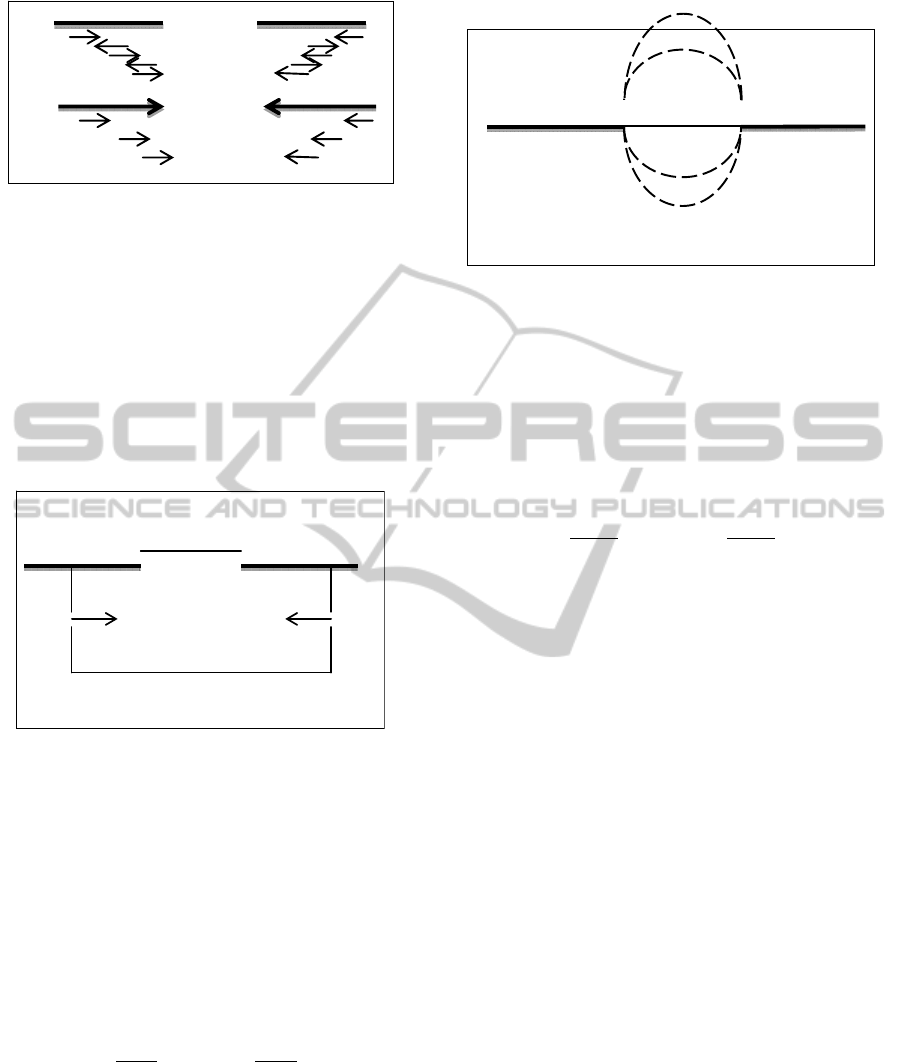
Figure 2: (a) Five mate-pairs are aligned to contig 1 and
contig 2. Out of five mate-pairs, three support forward
contig orientation of contig 1 and two support reverse
orientation. (b) After filtering, the two mate-pairs
supporting reverse orientation for contig 1 are removed
and both contigs are given orientation based on the
majority of mate-pairs.
3.4.4 Distance Calculation
The distance between contigs is calculated using
mate-pair links. Each edge distance is given weight
= 1.
Figure 3: D = m – (l – F(i)) – R(j) where D is the distance
between contigs, m is mean length of fragments in the
library, l is length of contig 1, F(i) and R(j) are respective
positions of alignment between the contig and read.
3.4.4.1 Edge Bundling
If there is more than one-mate pair link between a
pair of contigs, these mate-pairs are bundled into a
single distance provided that mate pair distances lie
in ± 3σ range from the median distance between
contigs. The length of the new edge after bundling is
p/q and standard deviation = 1/ √q where:
=
∑
and =
∑
(1)
This process is repeated until no edges fall in this
range. The weight of the new edge is equal to the
sum of the weights of the bundled edges. (Huson,
2002).
Figure 4: D1, D2, D3, D4 are distances between a pair of
contigs with different mate pairs. D represents distance
between contigs after edge bundling.
3.4.4.2 Weighted Reduction
If there are still edge distances which cannot be
bundled using the above criterion, we perform
weighted bundling, taking the weight of all edges
into account:
l
e
=
∑
∑
and
=
∑
∑
(2)
Where l(e) denotes length of the new edge and σ(e)
denotes the standard deviation of the new edge. The
weight of the new edge will be the sum of the
weights of all merged edges.
3.4.5 Contig Overlap Graph
A contig overlap graph is created for a given set of
contigs. Each node represents a contig and contigs
are connected to other contigs if there is k -1
overlap, where k denotes kmer length used for
construction of contigs. (Huson, 2002).
3.4.6 Graph Traversal
The graph is traversed in a depth-first search manner
to look for a single unique path from start contig C
i
until all contigs paired with C
i
are included in the
path. As the graph can be extremely dense in
repetitive areas, parameter threshold value is defined
to limit the depth of search in the graph. This
process is repeated for each contig C
i.
The final step
removes overlapping contigs and stitches together
the consistent paths to generate the scaffolds.
(
b
)
(
a
)
Contig 1
Contig 2
Mean and standard deviation of library (m, σ)
Distance D
F(i)
R(j)
Distance D
1
Conti
g
1
Conti
g
2
D
istance D
3
Distance D
2
Distance D
4
Distance D
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
200
4 PARALLELIZATION
IN PADENA
Each step in PadeNA is individually parallelized
which aids in extensibility and reusability of code.
4.1 Parallel de Bruijn Graph
Construction
A de Bruijn Graph is the core data structure used in
the assembly process. In PadeNA, we have
developed a unique implementation of the de Bruijn
Graph for shared memory architecture systems. The
entire read set is portioned using .NET Partitioner
equal to the number of cores in the system. Then
individual core constructs kmer dictionary on
portioned reads. Finally, dictionaries are merged into
a single dictionary. While constructing dictionary
forward and reverse complement are treated as
same. Each kmer sequence as key in dictionary is
defined as a node of a de Bruijn Graph. The
adjacency information of each node is generated
independently. Each node can be connected to a
maximum of 8 neighbors, each sharing a (k -1)
overlap with the node either in the forward or
reverse direction. This connectivity information is
also stored in nodes to fasten the step of graph
traversal.
4.2 Error Removal
Dangling links identification and removal steps are
both performed as a parallelized activity. Redundant
paths which are also present can be similarly
removed in parallel. For definitions of dangling links
and redundant paths, please refer to the assembly
algorithm section.
4.3 Scaffold Generation
Scaffold generation is the second phase of assembly.
As with previous phase of assembly, each step is
individually parallelized.
4.3.1 Contig Overlap Graph
The contig overlap graph is a core data structure for
the second phase of assembly. Each contig is
considered as a node of the graph. Each contig
independently locates its neighbor such that each
neighbor should have a (k-1) overlap in either
forward or reverse complement direction.
4.3.2 Depth-First Search
The contig overlap graph is traversed in a depth-first
fashion to generate all possible paths meeting the
distance constraints imposed by mate-pair data. This
step can be parallelized because each path
originating from different node can be traversed
independently and a list of paths generated. These
paths may then be merged to generate scaffolds.
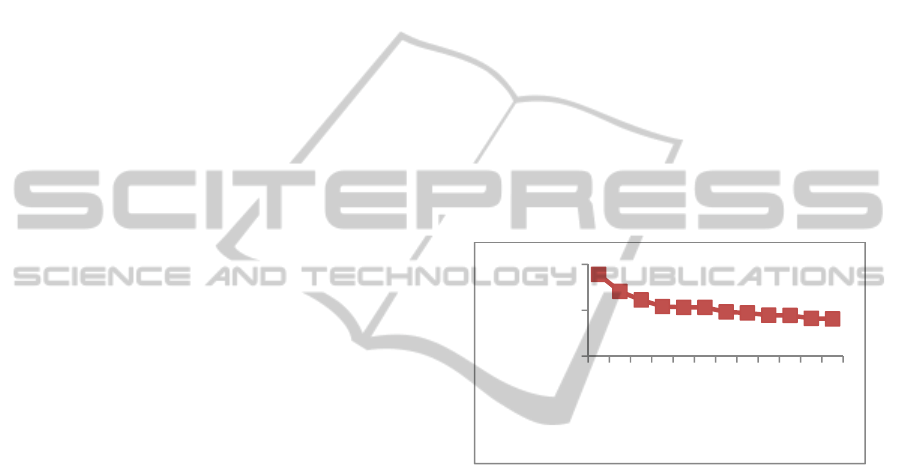
4.4 Scalability of PadeNA
on different cores
The scalability of the algorithm is a major concern
while parallelization of algorithm. The Euler dataset
as explained in results section is used to assemble
using default parameters on various system
configurations.
The scalability of the algorithm depends on size of
the data and error rate.
Figure 5: Variation of time taken for assembly vs. number
of processors.
5 ASSEMBLY ANALYSIS
For all data sets, only contigs ≥ 100 bp in length
were used for evaluation. In addition, contigs are
only considered to be aligned, if they align ≥ 95% to
a reference genome, if available. The parameters
used to estimate quality of assembly are:
• N50: N50 is a statistical measure given in base
pairs, such that 50% of the assembled genome
lies in contigs of at least this length.
Genomic Coverage: The percentage of bases
of reference genome covered by contigs or
scaffolds. This is computed using the MUMmer
(Kurtz, 2004) tool where a reference genome is
available.
• Largest contig/Mean size of contigs/number
of contigs ≥ 100 bp
These parameters are calculated and used as a
guideline to denote relative quality of the
0
50
100
24681012141618202224
Time (in seconds)
Number of Cores
PadeNA: A PARALLEL DE NOVO ASSEMBLER
201
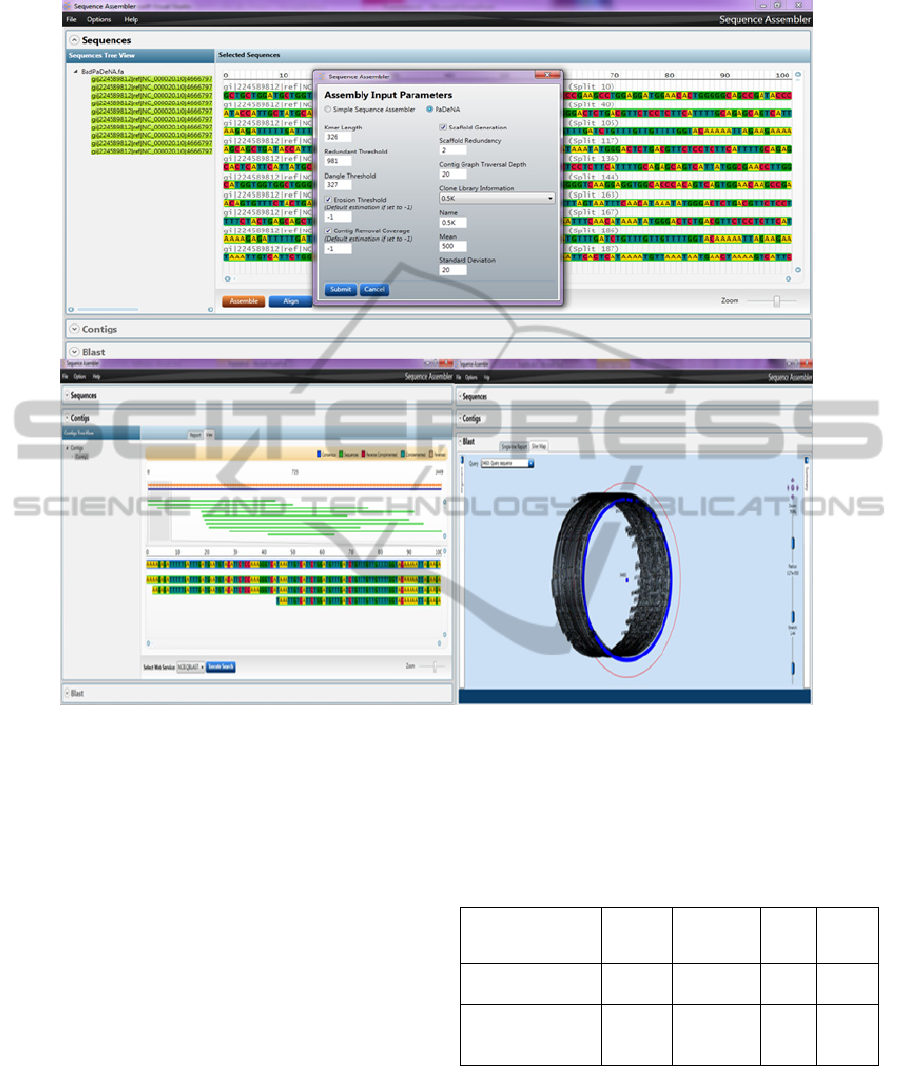
Figure 6: Sequence assembler view of sequence assembly and BLAST result for assembled sequence.
assembly performed (De Novo, 2009). MUMmer is
used for alignment because of its speed and
suitability for genome-level alignments
6 RESULTS
6.1 Evaluation of PadeNA Assembly
using Euler Data
The data used for the analysis is the data provided
by the Euler SR (Chaisson, 2008) tool as test data. It
is a 6.8 MB paired-read dataset with mean insert
length of 1000bp and standard deviation of 500bp.
We performed assemblies with Euler SR. version
1.1.2 and PadeNA version 1.0. Assemblies were
analyzed using the above-mentioned assembly
analysis parameters.
The results show PadeNA produces more
contigs, which may be due to repeats in the base
sequence. The reference sequence was not available
and so we were not able to calculate genomic
coverage and analyze the reason for a large number
of contigs.
Table 1: Comparison of PadeNA output quality against
Euler SR.
Assembler
Contigs
≥ 100 bp
Mean Size
(in bp)
N50
Largest
Contig
(in bp)
Euler SR version
1.1.2 (k = 20)
19 5185 14335 30523
PadeNA
(k = 20 and Depth =
20
50 10543 30628 30673
7 DISCUSSION
The PadeNA algorithm described above is included
as part of the broader Microsoft Biology Foundation
(MBF) library of general bioinformatics
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
202

functionality, and is available directly to the
application developer or may be accessed by the
user via the included Sequence Assembler
demonstration application (Fig. 6). The Sequence
Assembler application is a GUI-based interface to a
range of MBF functions and uses rich user interface
elements to enable visualization and manipulation of
genomic data. The user can perform assembly,
alignment and multiple sequence alignment of DNA,
RNA and protein sequences, visualizing the output
in a graphical alignment display built using the
Windows Presentation Foundation and Silverlight.
The Sequence Assembler also provides a connector
to various BLAST (Altschul, 1997) web services,
which can be used to characterize an assembled
sequence using public databases.
While our initial results are promising, some work is
needed to further improve the quality and utility of
the assembled output, especially for large size
genomes. Nonetheless, PadeNA can currently be
used for assembling bacterial genomes on shared
memory architectures and each step can be
customized to handle datasets with different
characteristics, or better meet the needs of different
groups of scientific users.
ACKNOWLEDGEMENTS
We would like to thank the Aditi-Microsoft MBF
Engineering team for their continued support to
make this de novo assembler design and technical
implementation deep, robust and of very high
quality. We would also like to thank Steve Jones,
Inanc Birol and other staff at Canada’s Michael
Smith Genome Center for their kind assistance in
understanding the field of genomics. Last but not
least, a very special thanks to Prasanth Koorma for
his constant motivation and encouragement
throughout the project.
REFERENCES
Altschul Stephen F., Madden Thomas L., Schaffer
Alejandro A., Zhang Jinghui, Zhang Zheng, Miller
Webb, & Lipman David J. 1997,’ Gapped BLAST and
PSI-BLAST: a new generation of protein database
search programs’, Nucleic Acids Res. 25:3389-3402.
Batzoglou S., Jaffe D.B., Stanley K., Butler J., Gnerre S.,
Mauceli E., Berger B., Mesirov J. P., & Lander E. S.,
2002, ‘ARACHNE: a whole-genome shotgun
assembler’, Genome Research, 12:177–189.
Biswas Surupa 2006, The Performance Benefits of NGen.,
Viewed July 5
th
2010, < http://msdn. microsoft.com/
en-us/magazine/cc163610.aspx>
Butler J., MacCallum I., Kleber M., Shlyakhter I. A.,
Belmonte M. K., Lander E. S., Nusbaum C. N., &
Jaffe D. B., 2008, ‘ALLPATHS: De novo assembly of
whole-genome shotgun microreads’, Genome
Research, 18:810–820.
Chaisson M.J. & Pevzner P.A., 2008, ‘Short fragment
assembly of bacterial genomes’, Genome Research,
pages 18:324–330.
De Novo Assembly using Illumina reads – technical note:
Illumina sequencing, 2009, retrieved July 5
th
2010,
<http://www.illumina .com/Documents/products/tech
notes/technote_denovo_assembly.pdf>
Green P., 1996, ‘Documentation for Phrap. Technical
report’ Genome Center, University of Washington.
Havlak P., Chen R., Durbin K. J., Egan A., & Ren Y.,
2003, ‘The atlas genome assembly system’, Genome
Research, 14:721–731.
Huang X. & Madan A., 1999, ‘CAP3: A whole-genome
assembly program’, Genome Research, 9:868–877.
Huson Daniel H., Reinert Knut, & Myers Eugene W.,
2002, ‘The greedy path-merging algorithm for contig
scaffolding’, Journal of the ACM (JACM) archive,
Volume 49, Issue 5.
Kurtz S., Phillippy A., Delcher A. L., Smoot M.,
Shumway M., Antonescu C., & Salzberg S. L., 2004,
‘Versatile and open software for comparing large
genomes’, Genome Biology.
Mono: Cross platform, open source .NET development
framework, 2004. Viewed July 5
th
2010, <
http://mono-project.com/Main_Page>
Myers E. W., Sutton G. G., Delcher A. L., & Dew I. M.,
2000, ‘A whole-genome assembly of Drosophila’,
Science, 287(5461):2196–2204.
Pattison Ted 1999, Understanding Interface-based
Programming, Viewed July 5
th
2010, <
http://msdn.microsoft.com/en-us/library/aa 260635
(VS.60).aspx>
Pevzner P. A., Tang H., & Waterman M. S., 2001, ‘An
eulerian path approach to DNA fragment assembly’,
Proceedings of the National Academy of Sciences,
98(17):9748–9753.
Pop M., Kosack D. S., & Salzberg S. L., 2004,
‘Hierarchical scaffolding with Bambus’, Genome
Research, 14 (1), pp. 149-159.
Simpson J. T., Wong K., Jackman S. D., Schein J. E.,
Jones S. J., & Birol I., 2009, ‘ABySS: A parallel
assembler for short read sequence data’, Genome
Research.
Sutton G. G., White O., Adams M. D., & Kerlavage A. R.,
1995, ‘TIGR assembler: A new tool for assembling
large shotgun sequencing projects’, Genome Science
and Technology, 1:9–19.
Zerbino D. & Birney E., 2008. ‘Velvet: Algorithms for de
novo short read assembly using de Bruijn graphs’,
Genome Research, 18:821–829
PadeNA: A PARALLEL DE NOVO ASSEMBLER
203
