
APPLYING CONCEPTUAL MODELING TO ALIGNMENT TOOLS
ONE STEP TOWARDS THE AUTOMATION
OF DNA SEQUENCE ANALYSIS
Maria Jos´e Villanueva, Francisco Valverde and Oscar Pastor
Centro de Investigaci´on en M´etodos de Producci´on de Software, Universidad Polit´ecnica de Valencia
Camino de Vera S/N Valencia, Spain
Keywords:
Variation detection, Alignment tools, Software engineering.
Abstract:
Nowadays, the search of variations in DNA samples according to a reference sequence is performed using
several bioinformatic tools. Due to the process complexity, none of these tools fulfill all the functionality
required by biologists. For that reason, the definition of an integration process between these different tools
becomes a mandatory requirement. One interesting issue is that bioinformatic tools do not comply with any
standard format for expressing the output reports. As a consequence, the flow among tools must be manually
solved. This paper proposes a conceptual model in order to formalize how the output from alignment tools
must be produced. This work also provides a textual format based on this conceptual model. Thanks to both
contributions, the integration is handled in the problem space and the related technological details are avoided.
As a proof of concept of these ideas, the proposed format has been applied in a DNA sequence analysis process
which uses two bioinformatic tools.
1 INTRODUCTION
DNA sequence analysis is a process that is currently
not efficiently solved in the context of disease diag-
nosis. Because of the complexity of the process, sev-
eral different tools are required to produce an accu-
rate analysis. Biologists claim that none of these tools
provide all the functionality required to fulfill a com-
plete sequence analysis process (Rusk, 2009). Briefly,
this process is divided into several phases that are per-
formed with a different tool or by the biologist:
1. Basecalling phase: basecalling tools obtain the
nucleotide chain from an electropherogram.
2. Basecalling revision phase: biologists correct the
sequence provided by the basecalling tools.
3. Variation detection phase: alignment tools obtain
the variations of a sequence regarding a reference
sequence.
4. Phenotype assessment phase: variation analysis
tools associate the suitable phenotype to every
variation found.
5. Diagnosis report phase: biologists gather manu-
ally all the results and write down their conclu-
sions in a report.
In order to support the whole analysis process, all
these different tools and manual procedures must be
combined. The main drawback of this approach is
that the data flow among them is not a trivial task that
must be performed manually for every analysis. Con-
cerning variation detection (3) and phenotype assess-
ment phases (4), an automatedintegrationof tools still
cannot be achieved because of: 1) the lack of stan-
dards in the output results of alignment tools; and 2)
the ambiguity about what exactly has to be imported
by variation analysis tools.
This work proposes a solution for both problems
in order to support the integration between align-
ment tools and variation analysis tools. The presented
approach introduces the use of conceptual models
(K¨uhne, 2005) to provide a formal definition about
what exactly a variation is and, which are the relevant
concepts in a variation report.
With the aim of formalizing variation reports, this
work reviews several alignment tools used by biolo-
gists in the variation detection phase. This review has
been useful to determine which kind of variations are
detected and how they are usually described. From
the extracted conclusions, a conceptual model is de-
fined to support the formal specification of these vari-
137
Villanueva M., Valverde F. and Pastor O..
APPLYING CONCEPTUAL MODELING TO ALIGNMENT TOOLS ONE STEP TOWARDS THE AUTOMATION OF DNA SEQUENCE ANALYSIS.
DOI: 10.5220/0003142001370142
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2011), pages 137-142
ISBN: 978-989-8425-36-2
Copyright
c
2011 SCITEPRESS (Science and Technology Publications, Lda.)

ations reports. Based on this conceptual model, a tex-
tual format is defined using the XMLSchema speci-
fication (Biron and Malhotra, 2004). For validation
purposes, this textual format has been applied into the
integration between two alignment tools and one vari-
ation analysis tool.
The rest of the paper is organized as follows. In
section 2, the related work is presented. Section 3 ex-
plains the alignment tools review. Section 4 presents
a conceptual model that formalizes the variation re-
ports and the proposed XML format. In section 5,
the contributions of this work are applied into a real
integration scenario. Finally, section 6 presents the
concluding remarks.
2 RELATED WORK
In order to formalize the heterogeneity of the con-
cepts in the genomic domain, several works propose
the use of conceptual models. For instance (Paton
et al., 2000) describes a collection of conceptual mod-
els in yeast. Paton’s work models general genomic
data and data related to experiments, proteins or alle-
les. Another approach, as PaGE-OM (Brookes et al.,
2009), proposes a conceptual model that represents
genomic data in relation to assays performed by biol-
ogists. The project Atlas (Shah et al., 2005) presents
an integration attempt that defines the genomic data
models to be integrated from different databases. And
finally, the Gene Ontology (Gene Ontology Consor-
tium, 2004) defines a set of vocabularies and classifi-
cations, which are related to biological functions, pro-
cesses, and cellular components.
A common issue in these approaches is that the
proposed conceptualizations are highly related to the
experimental data, the used technologies or the rep-
resentation formats. As a consequence, these ap-
proaches cannot be easily adopted by variation analy-
sis tools. The purpose of this work is to use concep-
tual models to achieve a domain representation that
only considers the precise biological concepts.
Focusing on the problem of alignment output rep-
resentation, other attempts to solve the lack of stan-
dards can be found as well. For example, the Se-
quence Alignment Map (SAM) format (Li et al.,
2009) is a compact format to express variation results
from alignments. The main drawbacks are the com-
plexity of the syntax and the mandatory implemen-
tation of a low level mechanism to extract the data.
Our proposal overcomes these drawbacks by the use
of a conceptual model that is easier to understand by
biologists. The complexity of data representation is
reduced thanks to the formalization of the variation
detection domain. As one implementation of this con-
ceptual model, it is presented a textual format based
on the XML language: a standard language supported
by several software development environments. The
implementation of the software integration compo-
nents is simplified by the conceptual model and the
corresponding XML format, that can be used inside a
model-driven software development process.
3 ALIGNMENT TOOLS REVIEW
With the purpose of detecting the most relevant con-
cepts that alignment tools use in their reports, a set
of the most representative ones has been reviewed:
Sequencher (Gene Codes Corporation, 2010), SeqS-
cape (Applied Biosystems, 2010), Mutation Surveyor
(Softgenetics, 2010), Codon Code Aligner (Codon
Code Corporation, 2010), Polyphred (Department
Genomic Sciences, 2010), InSNP (Manaster et al.,
2005) and the WebTool BLAST from the NCBI
(NCBI, 2010).
To perform this review a real test has been carried
out with these tools. Real samplesof the BRCA1 gene
were provided by a research laboratory to give value
to the results. The strategy followed in this test was:
1. Installation of the tools in a computer under
Windows 7 (Sequencher, SeqScape, Mutation-
Surveyor, CodonCode Aligner, old versions of
Staden and InSNP). For the tools only supported
in Linux, the installation was done in another
computer under Ubuntu v8.04 (Polyphred).
2. Reading of the introduction tutorials and user
guides to understand the general principles of the
tool, the graphical user interface and the sup-
ported functionality.
3. Checking of the functionality for each tool, using
the samples provided.
4. Searching of variations within the samples in or-
der to compare the results and limitations under
the same conditions.
While working with these tools, the required concepts
around variations have been gathered and three main
issues have been detected: In the first place, the in-
troduction of a complete DNA Sequence is not pos-
sible due to technological limitations. Sequencing
machines are constrained to a maximum sequence
length, so the sequenced region must be split up in
small pieces called contigs. In the second place, the
limitations of the sequencing process produces erro-
neous bases. So, in order to improve the analysis
quality, this process must be realized several times.
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
138

Table 1: Alignment tools comparison.
Sequencher SeqScape CCAligner M.Surveyor Polyphred InSNP Blast
Sample edition X X X x x x x
Assembly
among samples X X X x x x X
to RefSeq X X X X X X X
Variations
Homozygosis
insertions X X X X x x X
deletions X X X X x x X
indels X X X X X X X
Heterozygosis
insertions x X X X x X -
deletions x X X X x X -
indels X X X X X X -
Report Format
PDF X X X X x x x
TXT X X X X X X X
XLS X X X X x x x
XML x X x X X x X
HTML X x x X x x x
From all the sequences obtained, a consensus se-
quence is derived. And, in the third place, some vari-
ations can not be expressed with the common letters
used to identify the DNA bases. Due to the fact that
a DNA sequence is made up of two alleles, varia-
tions can be homozygous, if the nucleotide changes
in both alleles, or heterozygous, if the nucleotide only
changes in one allele. As a consequence, an addi-
tional set of specific letters is required for reporting
heterozygous variations.
Concerning the functionality of the tools, the gen-
eral procedure workflow is defined in five steps:
1. Alignment project creation: the contigs to be an-
alyzed are introduced into the tool and the align-
ment is configured according to several parame-
ters.
2. Contigs Assembly: the different contigs are or-
dered and aligned according to a reference se-
quence.
3. Contig basecalling correction: Biologists check
the different contig bases using as guideline the
reference sequence and their knowledge in the
field. Then the errors produced by the sequenc-
ing machine or the basecalling algorithms are cor-
rected and a consensus sequence is obtained.
4. Comparison: An alignment between the consen-
sus sequence and a reference sequence is carried
out to search for variations (insertions, deletions
and indels in homozygosis or heterozygosis).
5. Report generation: All the detected variations are
gathered in a variation report. This report can be
exported and used in another bioinformatic task,
for instance to document which variation can pro-
duce a disease.
A comparison among all tools is summarized the
Table 1. According to these results, all tools are able
to assembly sequences into its correct position in-
side a reference, but only Sequencher, SeqScape and
Codon Code Aligner support the sample edition to
correct basecalling. Regarding variation detection Se-
qScape, Codon Code Aligner and Mutation Surveyor
are the only tools that search for all kinds of varia-
tions. Each tool uses its own notation to generate the
reports and several formats to export these reports.
4 CONCEPTUAL MODEL FOR
VARIATION REPORTS
The main contribution of this work is to formalize the
common concepts that are used in the alignment tools
for generating the output reports. Taking into account
the common expressiveness from these tools, a con-
ceptual model has been defined (Figure 1).
While performing the variation detection phase,
the first step is to align the input sequence and a ref-
erence sequence. One Alignment is always defined
by a Consensus sequence and a Reference sequence.
Both conceptual entities inherit from the conceptual
APPLYING CONCEPTUAL MODELING TO ALIGNMENT TOOLS ONE STEP TOWARDS THE AUTOMATION OF
DNA SEQUENCE ANALYSIS
139
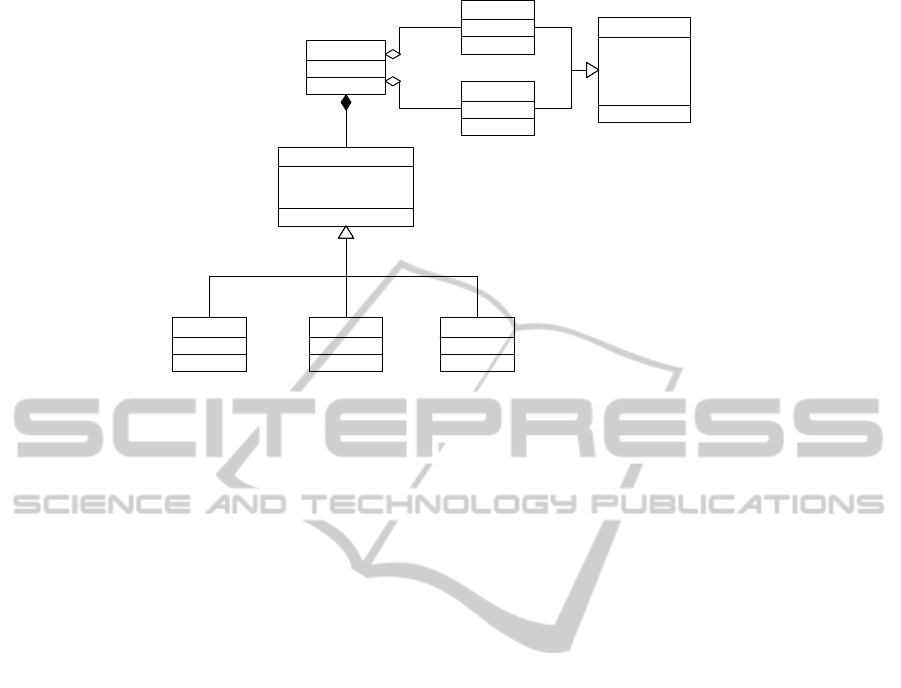
-geneId : string
Alignment
-startPos : int
-endPos : int
-isHeterozygous : bool
Difference
-bases : string
Insertion
-length : int
Deletion
-bases : string
Substitution
Reference
Consensus
1
*
1..*
1
1..*
1
-id : int
-refSource : string
-sequence : string
-startPos : int
-endPos : int
DNASequence
Figure 1: Alignment Report Conceptual Model.
entity DNASequence. The Alignment entity has an at-
tribute called geneId, which identifies the gene to be
analyzed. For standardization purposes, this attribute
complies with the standard nomenclature of Human
Genome Nomenclature Committee (HGNC) (Povey
et al., 2001)
A DNASequence defines the set of features associ-
ated to a sequenced DNA sample. A DNASequence is
represented by a numerical identifier, a sequence that
is a string of letters representing the nucleotides of
the sample, a refSource that indicates the datasource
(a database, a local file, etc.) where the sequence
comes from, and a range composed by startPos and
endPos sequence positions, that can be used to es-
tablish a delimitation in the sequence. The Consen-
sus entity models the DNA sequence that is analyzed,
for instance a patient sample, and the Reference entity
models a DNA sequence usually used for comparison
purposes.
All the differences found in the Alignment be-
tween both sequences are considered variations and
are modeled by the entity Difference. When a vari-
ation is found in one Alignment, the position where
it is located has to be indicated. To avoid ambigu-
ities, and following the recommendations of Human
Genome Variation Society (HGVS) (Den Dunnen and
Antonarakis, 2000), the Difference entity has two at-
tributes for defining where a variation starts and ends:
startPos and endPos. Moreover a Difference has the
boolean attribute isHeterozygous that indicates if a
variation occurs in one allele or in both alleles (ho-
mozygosis).
Differences are categorized into three entities ac-
cording to the change performed in the sequence:
Insertion (additional nucleotides are inserted), Dele-
tion (several nucleotides are deleted), and Substitu-
tion(some nucleotides change their value). The en-
tity Insertion has the attribute bases to indicate the in-
serted nucleotides; the entity Deletion has an attribute
length to indicate how manynucleotides have been re-
moved and the entity Substitution has also an attribute
bases that indicates the new value of the changed nu-
cleotides.
The presented conceptual model is implemented
defining a corresponding XMLSchema. An example
of this XML format is:
<alignment geneId="NF1">
<Reference id="Ref001">
<refDB>NG_009018.1</refDB>
</Reference>
<Consensus id="Query001">
<sequence>atggta....aattggcca</sequence>
</Consensus>
<Differences>
<Sub endPos="15" initialPos="16">c</Sub>
<Sub endPos="20" initialPos="20"
heterozygous="true">a </Sub>
<Del length="5" endPos="52"
initialPos="48">aaaaa</Del>
<Del length="1" endPos="68"
initialPos="68">g</Del>
<Ins endPos="77" initialPos="76">t</Ins>
</Differences>
</alignment>
5 PROOF OF CONCEPT:
SEQUENCE ANALYSIS TOOLS
INTEGRATION
Thanks to the conceptualization of the variation re-
ports, the data flow between alignment tools and
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
140

Scenario1
Sequencher
Scenario 2
PROS
Protototype
Blast NCBI
Specific
Translator A
Specific
Translator B
Sequencher
Blast NCBI
Conceptual
Model
Common
XML Schema
Variations
Report
Variations
Report
Variations
Report
Variations
Report
-geneId : string
Alignment
-startPos : int
-endPos : int
-heterozygous : bool
Difference
-bases : string
Insertion
-length : int
Deletion
-bases : string
Substitution
Reference
Consensus
1
*
1..*
1
1..*
1
-id : int
-refSource : string
-sequence : string
-startPos : int
-endPos : int
DNASequence
PROS
Protototype
Figure 2: Integration process.
variation analysis tools becomes a systematized step.
Concretely, the integration between these two type of
tools can be easily implemented using the proposed
XML format.
At the moment, data flow between alignment tools
and variation analysis tools requires the development
of format translators to achieve the communication
among tools (see Figure 2). Alignment tools gener-
ate reports in their own formats and variation analysis
tools import data also in their own formats. Hence,
the integration requires a specific translator to trans-
form the format of each alignment tool to the for-
mat of each variation analysis tool (Scenario 1). The
problem lays in the fact that these translators are not
reusable. So the more tools to be integrated the more
translators must be implemented.
However, this work solves the dependency among
tools and reduces the number of translators using the
conceptual model. As each tool manages the same
concepts (already defined in the conceptual model),
the integration is achieved by means of using the same
XML format (Scenario 2). On the one hand, the re-
ports generated by alignment tools must be expressed
following the conceptual model. Therefore, each re-
port format is translated to the common XML for-
mat. On the other hand, variation analysis tools read
data from the conceptual model, so XML data is con-
verted to each input format. Using this solution the
developed translators can be reused in other integra-
tion process. For this reason it is only necessary to
develop one translator for each tool to be integrated,
for alignment tools and variation analysis tools.
For evaluation purposes, the alignment tools Se-
quencher and Blast from the NCBI Website have been
integrated with a variation analysis tool. The selected
tool is the PROS Prototype Tool (Martinez et al.,
2010). Hence, three translators are implemented:
1. In the case of Sequencher, a translator that obtains
the reference sequence, the consensus, the varia-
tions, and creates the XML file.
2. In the case of the BLAST Webtool, a transla-
tor that obtains the reference, the consensus se-
quence, parses the output to obtain the variations
and creates the XML file.
3. Regarding the PROS prototype, since it is devel-
oped in Java language, it has been used the JAXB
(Java Architecture for XML Bindings) API (Ort
and Mehta, 2003). This API allows the parsing
of XML data into objects available in the con-
text of the application. In order to consume the
XML data, the translator instantiates the classes
obtained with the binding compiler of JAXB (xjc),
extracts the data required and transforms it into
objects that can be used by the variation analysis
tool.
Because of the three translators implementation, the
flow among the tools is supported. Therefore, biolo-
gists perceive that the variation detection and the vari-
ation analysis phases are executed in a single step.
6 CONCLUDING REMARKS
This work proposes a conceptual model to achievethe
integration of biological tools that perform two differ-
APPLYING CONCEPTUAL MODELING TO ALIGNMENT TOOLS ONE STEP TOWARDS THE AUTOMATION OF
DNA SEQUENCE ANALYSIS
141

ent phases of a DNA sequence analysis process.
The use of this conceptual model as a integra-
tor solution provides several advantages in relation
to the current state: On the one hand, the conceptual
model is based on the common biological concepts
used by the alignment tools. Furthermore, because
the proposed implementation of the conceptual model
is based on the standard XML language, the data ex-
change among different processes and tools is feasi-
ble. On the other hand the use of conceptual models
provide several advantages: 1) concepts are well de-
fined; and 2) it is easier to reflect new changes and
adapt the software to the new requirements. If bio-
logical concepts change or alignment tools evolve, the
conceptual model and its implementations can be eas-
ily modified in order to reflect the new concepts. For
these reasons, biologists are free to choose the most
suitable alignment tool that fits their needs.
Apart from the benefits that offers this proposal, it
must be taken into account that it also presents several
issues: One issue is that the conceptual model could
be incomplete because the commercial tools has been
tested in trial versions, where some functionality is
restricted. Therefore it is possible that some concepts
are missing. Another issue arises because it is not
possible to modify the specific implementation of the
alignment tools. The data has to be previously ex-
ported in order to be translated to the proposed for-
mat. This additional step must be carried out by biol-
ogists, so the process is not fully automated. As fu-
ture work, with the goal of achieving a complete au-
tomation of DNA sequence analysis, there are some
phases that must be addressed as well. For instance,
the next step is to study how to create diagnosis re-
ports automatically taking into account the phenotype
associated to the reported variations.
ACKNOWLEDGEMENTS
Thanks to the Instituto de M´edicina Gen´omica
(IMEGEN, http://www.imegen.es) for its support pro-
viding the data. This research work is supported by
the Spanish MICINN under the FPU grant AP2010-
1985.
REFERENCES
Applied Biosystems (2010). Seqscape. http://www3.app
liedbiosystems.com/ABHome/index.htm.
Biron, P. V. and Malhotra, A., editors (2004). XML Schema
Part 2: Datatypes. W3C Recommendation. W3C, 2nd
edition.
Brookes, A. J. et al. (2009). The Phenotype and Geno-
type Experiment Object Model (PaGE-OM): A Ro-
bust Data Structure for Information Related to DNA
Variation. Human Mutation, 30(6):968–77.
Codon Code Corporation (2010). Codon Code Aligner.
http://www.codoncode.com/aligner/.
Den Dunnen, J. T. and Antonarakis, S. E. (2000). Muta-
tion Nomenclature Extensions and Suggestions to De-
scribe Complex Mutations: A Discussion. Human
Mutation, 15(1):7–12.
Department Genomic Sciences (2010). Polyphred.
http://droog.gs.washington.edu/polyphred/.
Gene Codes Corporation (2010). Sequencher.
http://www.genecodes.com/.
Gene Ontology Consortium (2004). The Gene Ontology
(GO) Database and Informatics Resource. Nucleic
Acids Research, 32(suppl1):D258–261.
K¨uhne, T. (2005). What is a Model. In Language Engineer-
ing for Model-Driven Software Development, number
04101 in Dagstuhl Seminar Proceedings, pages 200–
0. IBFI, Schloss Dagstuhl, Germany.
Li, H. et al. (2009). The Sequence Alignment/Map Format
and SAM Tools. Bioinformatics, 25(16):2078–2079.
Manaster, C. et al. (2005). InSNP: a tool for automated de-
tection and visualization of SNPs and InDels. Human
mutation, 26(1):11–19.
Martinez, A. M. et al. (2010). Facing the challenges of
genome information systems: a variation analysis pro-
totype. Caise Forum.
NCBI (2010). BLAST (Basic Local Alignment Search
Tool). http://blast.ncbi.nlm.nih.gov/Blast.cgi.
Ort, E. and Mehta, B. (2003). Java Architecture for XML
Binding (JAXB). Technical Report Sun Developer
Network.
Paton, N. W. et al. (2000). Conceptual modelling of ge-
nomic information. Bioinformatics, 16(6):548–57.
Povey, S., Lovering, R., Bruford, E., Wright, M., Lush, M.,
and Wain, H. (2001). The HUGO Gene Nomenclature
Committee (HGNC). Human Genetics, 109(6):678–
680.
Rusk, N. (2009). Focus on Next-Generation Sequencing
Data Analysis. Nature Methods, 6(11s):S1.
Shah, S. et al. (2005). Atlas - A Data Warehouse for Inte-
grative Bioinformatics. BMC Bioinformatics, 6(1):34.
Softgenetics (2010). Mutation Surveyor. http://www.soft
genetics.com/.
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
142
