
EXPERIMENTAL RESULTS ON MULTIPLE PATTERN MATCHING
ALGORITHMS FOR BIOLOGICAL SEQUENCES
Charalampos S. Kouzinopoulos, Panagiotis D. Michailidis and Konstantinos G. Margaritis
Parallel and Distributed Processing Laboratory, Department of Applied Informatics, University of Macedonia
156 Egnatia str., P.O. Box 1591, 54006 Thessaloniki, Greece
Keywords:
Algorithms, Multiple pattern matching, Multiple keyword matching, String searching, Biological sequence
databases.
Abstract:
With the remarkable increase in the number of DNA and proteins sequences, it is very important to study
the performance of multiple pattern matching algorithms when querying sequence patterns in biological se-
quence databases. In this paper, we present a performance study of the running time of well known multiple
pattern matching algorithms on widely used biological sequence databases containing the building blocks of
nucleotides (in the case of nucleic acid sequence databases) and amino acids (in the case of protein sequence
databases).
1 INTRODUCTION
Multiple pattern matching is the computationally in-
tensive kernel of many applications including infor-
mation retrieval, intrusion detection systems, web fil-
tering, virus scanners and spam filters. In recent years
however, an immediate interest in string-matching
problems as a powerful tool in locating nucleotide or
amino acid sequence patterns in biological sequence
databases has been witnessed. For example, when
proteomics data is used for genome annotation in a
process called proteogenomic mapping (Jaffe et al.,
2004), a set of peptide identifications obtained using
mass spectrometry is matched against a target genome
translated in all six reading frames.
The multiple pattern matching problem can be
defined as follows: Given a sequence database (or
text) T = t
1
t
2
...t
n
of length n and a finite set of r
patterns P = p
1
, p
2
, ..., p
r
, where each p
i
is a string
p
i
= p
i
1
p
i
2
... p
i
m
of length m over a finite character set
Σ, the task is to find all occurrences of any of the pat-
terns in the sequence database.
The naive solution to this problem is to per-
form r separate searches with a sequential algorithm
(Navarro and Raffinot, 2002). While frequently used
in the past, this technique is not efficient when a large
pattern set is involved. The aim of the multiple pattern
matching algorithms is to scan the input string T in a
single pass to locate the occurrences of all patterns.
These algorithms are based on single-pattern match-
ing algorithms, with some of their functions general-
ized to process multiple patterns simultaneously dur-
ing the preprocessing phase, generally with the use of
trie structures and hashing.
The multiple pattern matching is widely used in
computational biology for a variety of pattern match-
ing tasks. For example, Brundo and Morgenstern
use a simplified version of the Aho-Corasick algo-
rithm to identify anchor points in their CHAOS algo-
rithm for fast alignment of large genomic sequences
(Brudno and Morgenstern, 2002; Brudno et al., 2004).
Hyyro et al. demonstrate that Aho-Corasick outper-
forms other algorithms for locating unique oligonu-
cleotides in the yeast genome (Hyyro et al., 2005).
The SITEBLAST algorithm (Michael et al., 2005)
employs the Aho-Corasick algorithm to retrieve all
motif anchors for a local alignment procedure for ge-
nomic sequences that makes use of prior knowledge.
Buhler et al use Aho-Corasick to design simultaneous
seeds for DNA similarity search (Buhler et al., 2005).
This paper presents experiments for the running
time of the well known Commentz-Walter, Wu-
Manber, Set Backward Oracle Matching and Salmela-
Tarhio-Kyt¨ojoki multiple pattern matching algorithms
for biological sequences. A detailed analysis of the
multiple pattern matching algorithms presented in this
paper, additional experiments on different types of
data as well as a study on the preprocessing time and
the memory requirements of the algorithms can be
found in (Kouzinopoulos and Margaritis, 2010).
274
S. Kouzinopoulos C., D. Michailidis P. and G. Margaritis K..
EXPERIMENTAL RESULTS ON MULTIPLE PATTERN MATCHING ALGORITHMS FOR BIOLOGICAL SEQUENCES.
DOI: 10.5220/0003133202740277
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2011), pages 274-277
ISBN: 978-989-8425-36-2
Copyright
c
2011 SCITEPRESS (Science and Technology Publications, Lda.)

The aim of this experimental study is to identify a
suitable and preferably fast multiple pattern matching
algorithm for several problem parameters such as a
given biological database, the size of the pattern set,
and the length of the patterns.
2 EXPERIMENTAL
METHODOLOGY
The experiments were executed locally on an Intel
Core 2 Duo CPU with a 3.00GHz clock speed and 2
Gb of memory, 64 KB L1 cache and 6 MB L2 cache.
The Ubuntu Linux operating system was used and
during the experiments only the typical background
processes ran. To decrease random variation, the time
results were averages of 100 runs. All algorithms
were implemented using the ANSI C programming
language and were compiled using the GCC 4.4.3
compiler with the “-O2” and “-funroll-loops” opti-
mization flags.
To compare the pattern matching algorithms, the
practical running time was used as a measure. Practi-
cal running time is the total time in seconds an algo-
rithm needs to find all occurrences of a pattern in an
input string including any preprocessing time and was
measured using the MPI Wtime function of the Mes-
sage Passing Interface since it has a better resolution
than the standar clock() function.
The data set was similar to the ones used in (Sheik
et al., 2005) and (Kalsi et al., 2008). It consisted
of the SWISS-PROT Amino Acid sequence database
with a size of n = 182.116.687 characters and an al-
phabet of size 20, the FASTA Amino Acid (FAA)
sequence of the A-thaliana genome with a size of
n = 11.273.437 characters and an alphabet of size 20
and the FASTA Nucleidic Acid (FNA) sequence of
the A-thaliana genome with a size of n = 118.100.062
characters and an alphabet of size 4.
3 EXPERIMENTAL RESULTS
In this section, the performance of the algorithms is
evaluated according to their running time for different
biological databases.
Figures 1 to 3 present the running time of the algo-
rithms including preprocessing for the SWISS-PROT
amino acid sequence database and for the FASTA
amino acid and nucleidic acid databases of the A-
thaliana genome respectively for a pattern length of
m = 8 and m = 32 and for 100 to 100.000 patterns.
As can generally be seen from the Figures, by vary-
ing different parameters such as the size of the pattern
set, the length of the patterns and the size of the al-
phabet can affect the performance of the algorithms
in different ways.
In the case of the SWISS-PROT database and for a
pattern length of m = 8, the SOG and BG algorithms
had the best performance when up to 10.000 patterns
were used while the SBOM algorithm was faster for
more than 10.000 patterns. When a pattern length of
m = 32 was used, the SOG and BG algorithms had
the fastest running time for up to 20.000 patterns,
while SBOM was faster for more than 20.000 pat-
terns. The HG and Wu-Manber algorithms had an av-
erage performance for either m = 8 or m = 32 while
Commentz-Walter was consistently the slowest algo-
rithm in terms of running time.
For the FASTA amino acid database, and for a
pattern length of m = 8, the SOG and BG algo-
rithms were faster when up to 10.000 patterns were
used while for more than 10.000 patterns, the Wu-
Manber algorithm had the best performance, followed
by SBOM. When a pattern length of m = 32 was
used, the SOG and BG algorithms had the fastest
running time for up to 30.000 patterns. For bigger
pattern sets, Wu-Manber was the fastest algorithm.
Commentz-Walter was the algorithm with the worst
performance when m = 8 was used while for m = 32,
the Commentz-Walter and the SBOM algorithms had
the worst performance.
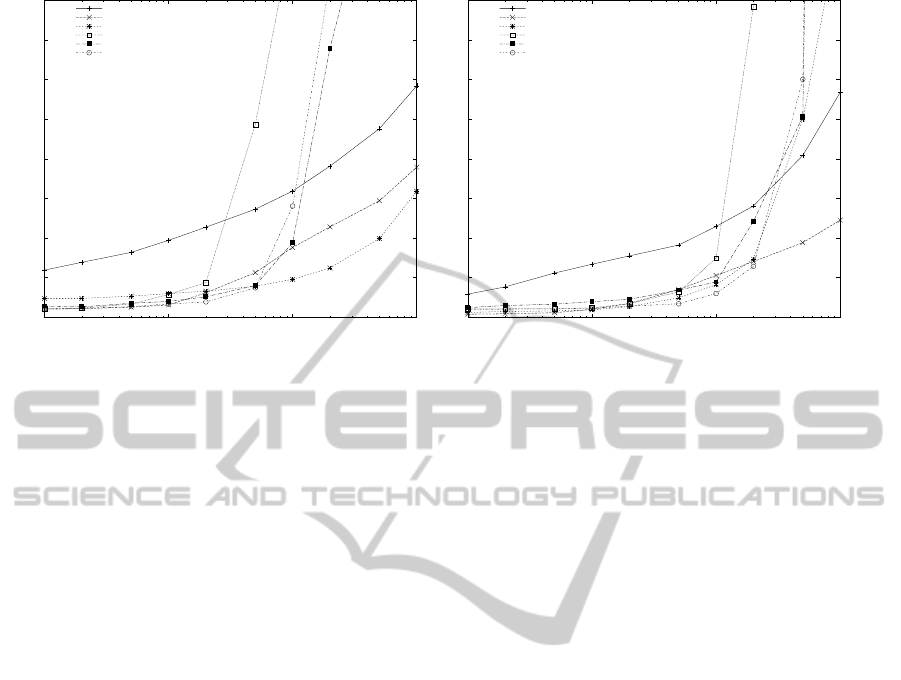
In the case of the FASTA nucleidic acid database,
SBOM was the algorithm that worked consistently
faster for a pattern length of m = 8. When up to 2.000
patterns were used, Commentz-Walter was the slow-
est algorithm while for more than 2.000 patterns, HG
was the algorithm with the worst performance. For
a pattern length of m = 32 the Wu-Manber was the
fastest algorithm, especially when more than 20.000
patterns were used.
Specific performancecomments on the algorithms
follow. Commentz-Walter was the algorithm with the
fastest running time when used on the FASTA nucle-
idic acid database with a pattern length of m = 32,
especially when more than 10.000 to 50.000 pat-
terns were used. The algorithm had the worst per-
formance when used on the SWISS-PROT and the
FASTA amino acid databases and thus its use is not
recommended in general on large alphabet sizes such
as amino acid databases. Wu-Manber was the fastest
algorithm on the FASTA amino acid database when
more than 10.000 patterns were used and on the
FASTA nucleidic acid for a pattern length of m = 32
together with the Commentz-Walter algorithm. On
the SWISS-PROT database and for a pattern length
of m = 8, the algorithm had a good performance with
EXPERIMENTAL RESULTS ON MULTIPLE PATTERN MATCHING ALGORITHMS FOR BIOLOGICAL
SEQUENCES
275

0
1
2
3
4
5
100 1000 10000 100000
Running time (sec)
Number of patterns (m=8)
CW
WM
SBOM
HG
SOG
BG
0
1
2
3
4
5
100 1000 10000 100000
Running time (sec)
Number of patterns (m=32)
CW
WM
SBOM
HG
SOG
BG
Figure 1: Running time of the algorithms for the SWISS-PROT Amino Acid database.
0
0.2
0.4
0.6
0.8
1
100 1000 10000 100000
Running time (sec)
Number of patterns (m=8)
CW
WM
SBOM
HG
SOG
BG
0
0.2
0.4
0.6
0.8
1
100 1000 10000 100000
Running time (sec)
Number of patterns (m=32)
CW
WM
SBOM
HG
SOG
BG
Figure 2: Running time of the algorithms for the FASTA Amino Acid database.
a running time close to that of the other algorithms.
The SBOM algorithm outperformed the rest of
the algorithms when the SWISS-PROT database was
used and for a pattern set size of more than 10.000 to
20.000 patterns. It had also the best performance on
the FASTA nucleidic acid text when a pattern length
of m = 8 was used. On the FASTA amino acid had
an average performance for m = 8 and the worst per-
formance comparing to the rest of the algorithms for
m = 32. Among the Salmela-Tarhio-Kyt¨ojoki algo-
rithms, SOG and BG had the best performance in
practice when used on the SWISS-PROT database to-
gether with the SBOM algorithm and on the FASTA
amino acid database for fewer than 5.000 to 20.000
patterns. On the FASTA nucleidic acid database the
Salmela-Tarhio-Kyt¨ojoki algorithms had the slowest
running time comparing to the rest of the algorithms
and so their used is not recommended on small alpha-
bet sizes such as DNA-type databases.
4 CONCLUSIONS
In this paper, experimental results of the well
known Commentz-Walter, Wu-Manber, SBOM and
the Salmela-Tarhio-Kyt¨ojoki algorithms were pre-
sented. The algorithms were compared in terms of
running time for the SWISS-PROT Amino Acid se-
quence database and the FASTA Amino Acid (FAA)
and FASTA Nucleidic Acid (FNA) sequences of the
A-thaliana genome and for sets of size between 100
and 100.000 patterns with a length of m = 8 and m =
32. The experimental study proved that no algorithm
is the best for all values of the problem parameters.
Instead it was shown that for different databases, dif-
ferent algorithms are preferable: Commentz-Walter
had the best performance on the FASTA nucleidic
acid database for more than 10.000 patterns. Wu-
Manber was the fastest algorithm for the FASTA
amino acid database for more than 10.000 to 50.000
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
276
0
1
2
3
4
5
6
7
8
100 1000 10000 100000
Running time (sec)
Number of patterns (m=8)
CW
WM
SBOM
HG
SOG
BG
0
1
2
3
4
5
6
7
8
100 1000 10000 100000
Running time (sec)
Number of patterns (m=32)
CW
WM
SBOM
HG
SOG
BG
Figure 3: Running time of the algorithms for the FASTA Nucleidic Acid database.
patterns. The Wu-Manber algorithm also outper-
formed the rest of the algorithms on the FASTA nu-
cleidic acid database for a pattern length of m = 32
and for more than 20.000 patterns. The SBOM algo-
rithm had the best performance on the SWISS-PROT
database for m = 8 and for more than 10.000 patterns
and on the FASTA nucleidic acid database, especially
when a pattern length of m = 8 was used. Among the
Salmela-Tarhio-Kyt¨ojoki algorithms, HG did not per-
form well on any biological database while the SOG
and BG algorithms had the fastest running time on the
SWISS-PROT database when up to 10.000 to 20.000
patterns were used, and on the FASTA amino acid
database when up to 10.000 to 30.000 patterns were
used.
Since biological databases and pattern sets are
usually inherently parallel in nature, the work pre-
sented in this paper could be extended with a focus
on the speed up of the existing algorithms when paral-
lel processed on traditional parallel architectures like
cluster environmentsand multicore systems as well as
on modern parallel systems like GPU architectures.
REFERENCES
Brudno, M. and Morgenstern, B. (2002). Fast and sensitive
alignment of large genomic sequences. In IEEE Com-
puter Society Bioinformatics Conference, volume 1,
pages 138–147.
Brudno, M., Steinkamp, R., and Morgenstern, B. (2004).
The chaos/dialign www server for multiple align-
ment of genomic sequences. Nucleic Acids Research,
32:41–44.
Buhler, J., Keich, U., and Sun, Y. (2005). Designing seeds
for similarity search in genomic dna. Journal of Com-
puter and System Sciences, 70(3):342–363.
Hyyro, H., Juhola, M., and Vihinen, M. (2005). On exact
string matching of unique oligonucleotides. Comput-
ers in Biology and Medicine, 35(2):173–181.
Jaffe, J., Berg, H., and Church, G. (2004). Proteoge-
nomic mapping as a complementary method to per-
form genome annotation. Proteomics, 4(1):59–77.
Kalsi, P., Peltola, H., and Tarhio, T. (2008). Comparison
of exact string matching algorithms for biological se-
quences. Communications in Computer and Informa-
tion Science, pages 417–426.
Kouzinopoulos, C. S. and Margaritis, K. G. (2010). Al-
gorithms for multiple keyword matching: Survey and
experimental results. Technical report.
Michael, M., Dieterich, C., and Vingron, M. (2005).
Siteblast–rapid and sensitive local alignment of ge-
nomic sequences employing motif anchors. Bioinfor-
matics, 21(9):2093–2094.
Navarro, G. and Raffinot, M. (2002). Flexible pattern
matching in strings: practical on-line search algo-
rithms for texts and biological sequences. Cambridge
University Press.
Sheik, S., Aggarwal, S. K., Poddar, A., Sathiyabhama,
B., Balakrishna, N., and Sekar, K. (2005). Analysis
of string-searching algorithms on biological sequence
databases. Current Science, 89(2):368–374.
EXPERIMENTAL RESULTS ON MULTIPLE PATTERN MATCHING ALGORITHMS FOR BIOLOGICAL
SEQUENCES
277
