
HOW MUCH BOVINE RHODOPSIN CRYSTAL STRUCTURE IS
USEFUL FOR MODELING HUMAN GPCRS?
β2-Adrenergic Receptor as a Test Case
Anwar Rayan, Mohamed Hegaze and Jamal Raiyn
QRC-Qasemi Research Center,Al-Qasemi Academic College, P.O.B. 124, Baka El-Garbiah 30100, Israel
Keywords: Data mining of GPCR database, Homology modeling, 3D-structure prediction.
Abstract: Availability of realistic models for human G-Protein Coupled Receptors (hGPCRs) will aid structure-based
drug design (SBDD), thus shortening the time period needed for drug development and minimizing cross-
reactivity of drugs with other hGPCRs. Many researchers have constructed models for hGPCRs with
homology modeling techniques based on the X-ray structure of bovine rhodopsin and recently to β2-
adrenergic receptor which are the only two GPCRs that have high resolution crystal structures. In this study,
we evaluate the usefulness of the bovine rhodopsin crystal structures for modeling hGPCRs by analysis of
large database of human G-protein coupled receptors that are members of family A (rhodopsin family). The
recently released structure of β2-adrenergic receptor was used as a test case for validation purposes of our
findings. From pair-wise sequence alignment of each of the receptors in the database to bovine rhodopsin,
we come to the conclusion that only for few hGPCRs, X-ray structure of rhodopsin could be used as a
template for modeling the trans-membrane domains (TMDs).The detailed analysis of the whole database
shows that in general, similarity to bovine rhodopsin is found more in the middle/endoplasmic part than in
the exoplasmic part. The shift in the cytoplasmic end of TMD-6 that has been seen in the recently released
crystal structure of β2-adrenergic receptor could be understood well from our bioinformatics study. On the
basis of our results from this research, we propose to regard specific parts from the endoplasmic domain of
the rhodopsin helices as appropriate template for constructing models of other GPCRs, while most of the
exoplasmic parts of GPCRs in this family require other techniques for their modeling, due to the low
sequence similarity between the receptors and rhodopsin in that region.
1 INTRODUCTION
G-protein coupled receptors (GPCRs) are membrane
embedded proteins that have a typical structural
topology: seven transmembrane helices (7TMH)
connected by intracellular and extracellular loops,
with an extracellular N-terminal and an intracellular
C-terminal (Gether, 2000). GPCRs derive their name
from their ability to recruit and to regulate the
activity of intracellular heterotrimeric G-proteins.
Their main role is to transfer (transduce) a signal
across the cell membrane. Such signals emerge from
interactions of GPCRs with extracellular agents,
which are highly diverse entities (e.g., ions, biogenic
Abbreviations: hGPCR (human G-Protein Coupled Receptor),
TMD (Trans Membrane Domain).
amines, nucleosides, lipids, peptides, proteins, and
even light). These agents are called “ligands” or
“agonists” and ligand binding is followed by a
change in the state of a GPCR to one with decreased
affinity to G-proteins. Thus, the meeting between
such agonists and GPCRs results in the conversion
of “extracellular events” to intracellular responses
(Nurnberg et al., 1995).
GPCRs are implicated in a very wide range of
body functions and processes, including
cardiovascular, nervous, endocrine, and immune
systems. Also, their involvement in many disease
conditions such as asthma, cardiovascular disease,
central nervous system disorders, pain and others
has been proven or suspected and they are
considered to be the single largest group of drug
targets. It has been estimated that GPCRs comprise
~45% of drug targets (Drews, 2000) and more than
291
Rayan A., Hegaze M. and Raiyn J. (2009).
HOW MUCH BOVINE RHODOPSIN CRYSTAL STRUCTURE IS USEFUL FOR MODELING HUMAN GPCRS? - β2-Adrenergic Receptor as a Test
Case.
In Proceedings of the International Conference on Bio-inspired Systems and Signal Processing, pages 291-298
DOI: 10.5220/0001542402910298
Copyright
c
SciTePress

50% of current drugs are directed to GPCRs (Nambi
and Aiyar, 2003).
The number of known GPCRs is in the
thousands, and many more are being discovered as a
result of recent advances in genomics and
proteomics. To be useful for drug design, structures
of these drug targets should be elucidated, in order
to employ them by methods of “Structure Based
Drug Design” (SBDD). The structural aspects of
GPCRs are however a source of constant debate in
recent years (Bissantz et al., 2003).
Direct experimental study of GPCR structures is
currently too complicated due to their native
membrane environment. Until November 2007,
only a single G-protein-coupled receptor, bovine
rhodopsin, has been studied by high-resolution
crystallography (Palczewski et al., 2000; Okada and
Nakamichi, 2004). β2-adrenergic receptor was the
second GPCR to solve and its structure revealed fair
similarity to the model obtained based on rhodopsin
as a template (Rasmussen et al., 2007; Cherezov et
al., 2007). The prospects for elucidating the
structures of other GPCR are not very high, and
await a major breakthrough. With no other structures
at hand, rhodopsin and/or β2-adrenergic receptor are
considered to be the prototypes of the main family of
GPCRs, of type A.
Due to the lack of experimental 3D-structures of
other GPCRs, one could hope to gain from
approximations based on molecular models. While
“ab initio” modeling is not practical yet for any
protein, “homology” or “comparative” modeling are
quite established methods (Rayan et al., 2000) and
are expected to be especially successful in the GPCR
subfamily A, that is considered to have the general
features of rhodopsin. Indeed, many GPCR
structures have been modelled recently, based on the
template of bovine rhodopsin/β2-adrenergic
receptors, by using its backbone coordinates and
adding the appropriate side chains of each sequence
(Eszter and Zsolt, 2008). Such “homology” or
“comparative” modeling of GPCRs has been aided
mainly by experimental information from point
mutations and other experimental resources
(http://www.gpcr.irg/7tm/, 2006). The length of
helices in the TMD remain similar in the modelled
GPCRs to those of the template rhodopsin, and loops
are not included in the template construction, except
in those rare cases where loop lengths are similar to
those of rhodopsin. But other approaches for
constructing models of GPCRs suggest that GPCRs
could differ in their structure from rhodopsin even
though their general features are similar (Oliveira et
al., 2002).
There are a few indications to justify such
deviations from the rhodopsin structure, in
constructing models for other GPCRs. A review by
Baker and Sali (Schacham et al., 2001) has shown
that a homology model for a protein at medium size
at least and with sequence identity of less than 30%
to the template crystal structure is unreliable. The
averaged sequence identity of bovine rhodopsin to
hGPCRs is less than 20%, meaning a homology-
based approach is unlikely to provide a reliable
structure to be used for making predictions. Others
in the community think that this “rule” is correct in
globular proteins and it is doubtful if this “rule”
could be extended to membrane proteins. Also, this
rule does not specify how identity should be
distributed along a sequence. As much as the GPCRs
super family is united by an overall structural
topology and an ability to recruit and regulate the
activity of G proteins, sequence identity between
super family members, even in the more conserved
transmembrane cores is too low. Significant
sequence conservation is found, however, within
several subfamilies of GPCRs. The subfamily of
rhodopsin-like GPCRs is by far the largest (more
than 85% of GPCRs) and is characterized by the
presence of some 35 (out of ~190) highly conserved
residue positions in the TMD, that may be involved
in binding and/or in activation (Baler and Sali,
2001).
The conserved positions along the TM sequences
constitute less than 20%. In contrast, the intracellular
and extracellular loops and the N- and C- terminals
of GPCRs vary in their lengths and therefore they
pose an alignment problem. Palczewski, K. and his
colleagues (Baldwin et al., 1997) via investigation of
sequence analysis of the TMD of GPCRs
demonstrated that “… the extracellular domain is the
least conserved, while GPCRs display considerable
conservation toward the endoplasmic side...” While
this is an important observation, it lacks specific
quantitative character. The conclusions of that study
concentrated on individual residue conservation and
on microenvironment conservation, and have thus
detected the most conserved residues in the TMD.
The authors concluded by suggesting that “It is
reasonable to speculate that the overall fold of these
receptors is highly conserved”. One of the
implications of that study are thus, that it is
reasonable to use the overall structure of rhodopsin
to model the TMD of other GPCRs.
Therefore, the question remains open, to what
extent is the structure of rhodopsin useful as a
template for constructing models of other GPCRs? A
quantitative measure of conservation in that family
BIOSIGNALS 2009 - International Conference on Bio-inspired Systems and Signal Processing
292

of GPCRs could be helpful for deciding upon the
exact parts of rhodopsin that could be used as
templates for such comparative modeling, and those
that should better be excluded. Should we use the
full extent of TM helices, some of the helices, or
stretches of sequences along helices? It was already
noticed earlier that endoplasmic parts of the TMD
are more conserved than exoplasmic parts (Baldwin
et al., 1997). But what are the quantitative aspects of
that conservation and how do they impinge on the
most important decision, which is - how much of the
rhodopsin structure may be used to model other
GPCRs?
Between the two extreme approaches, to use the
full crystallographic structure of the TMD of
rhodopsin or to employ none of it, we propose an
alternative. From our quantitative analysis, we
assign the parts of the structure of rhodopsin that
may be used as a template, and suggest to construct
the rest by other methods that allow deviations from
the crystal structure of the template.
2 METHODS
In this study, we hope to examine if there is a
quantitative basis for modeling the TMDs of
hGPCRs based on the X-ray structure of bovine
rhodopsin. A database of 951 rhodopsin like
hGPCRs were achieved from RAND
Biotechnologies Ltd company. They have used in-
house software called GPCR-scanner to screen the
protein database of human species composed of
63125 proteins (Ensembl human database). Trans-
membrane domains allocations and multiple
sequence alignments were performed by applying
Intelligent Learning Engine technology (Mirzadegan
et al., 2003) from RAND Biotechnologies Ltd
company. Some of the 951 receptors are identical in
the TMDs and differ only in length of the protein
sequence or in the rest of the structure – the two
terminals and/or the extacellular and/or the
intracellular loops. Higher similarity in sequences
means better chance to have close three-dimensional
structures and high confidence to obtain reliable
model for the query protein.
Sequence Alignments
Stretches of helical sequences for each of the
GPCRs have been determined by TMDs-Scanner
(Rayan and Raiyn, 2008), and were subsequently
aligned with those of rhodopsin in the crystal
structure. The length of each helix was imposed by
the rhodopsin template (TMDs) and is 194 residues
in total. No insertions or deletions were considered.
The calculation of cumulative similarity of
sequences to bovine rhodopsin or any other receptor
CC
l
is expressed by the average of conservation
scores for single sequences positions:
%100∗=
k
n
C
ij
ij
(1)
Where l is the number of amino acid positions in
the sequence of a helix in the TMD and Cj is the
score of the bovine rhodopsin/or of any other
receptor amino acid at position j and can adopt a
value of 1 if the residues are identical and a value of
0 if the residues are not identical. This score was
calculated in order to evaluate the similarity for the
seven TMDs separately as well as the lower
endoplasmic part (G-protein binding) and the upper
exoplasmic part (ligand binding) of the TMDs or
over certain windows along the helices.
Optimization of Windows’ Positions
After a window width was determined, the first
residue in the helix starts the window and the
identity percentage to rhodopsin was evaluated for a
certain hGPCR. The window was then shifted by
one amino acid all along the helix as well as the
other helices. The evaluation has been performed for
all the hGPCRs in our database. The analysis was
done in a few windows of widths between 7-14
residues. We have concentrated on the results of
windows of 11 residues, which are close to about
three such turns, respectively.
3 RESULTS AND DISCUSSION
Conserved Residues in the TMDs
Looking on the frequencies of individual residues in
particular positions along the TMDs (unpublished
data) reveals that large number of positions are
enriched with certain type of amino acid. Very low
variability in specific position contents could mean
importance in signal transduction pathway or in
structural fold. Those residues are mostly found in
the endoplasmic half of the TMDs or interacting
with the membrane or phospholipids head groups in
the edges of the membrane. The frequencies in some
case are different from those reported by Tara
Mirzadegan et al (Baldwin et al., 1997). For
example, in helix I, Gly20, Leu23 and Val24 were
HOW MUCH BOVINE RHODOPSIN CRYSTAL STRUCTURE IS USEFUL FOR MODELING HUMAN GPCRS? -
ß2-Adrenergic Receptor as a Test Case
293
found 79.2% instead of 68%; 50.2% instead of 60%
and 36.6% instead of 66%, respectively. Position 9
in helix II is occupied by Leu in 92.9% while the
other amino acid types are mostly very hydrophobic
like Ile, Met or Phe. This position could be
important to determine the height of the helix by
fixing this hydrophobic moiety in interaction with
the membrane. Position 16 is occupied in 43.9% by
Ser or Thr which properly interact with Trp from
helix IV. Basic residues are dominant in the first two
positions of helix IV and helix VII. Those residues
and others could play important role in determining
the orientation of the GPCR relative to the
membrane.
Entire Similarity in the TMDs of the hGPCRs
To check the entire similarity between all members
in our database, the receptors were clustered by
requiring that clusters should be dissimilar at least
by x% (with x ranging between 1-100). For
example, assume that the threshold for clustering is
x%, then, if receptor A has sequence identity with
receptor B less than the particular threshold, the two
receptors are considered one cluster.
How much human rhodopsin-like GPCRs are
similar?
0
200
400
600
800
10 30 50 70 90
Identity percentage threshold
Number of clusters
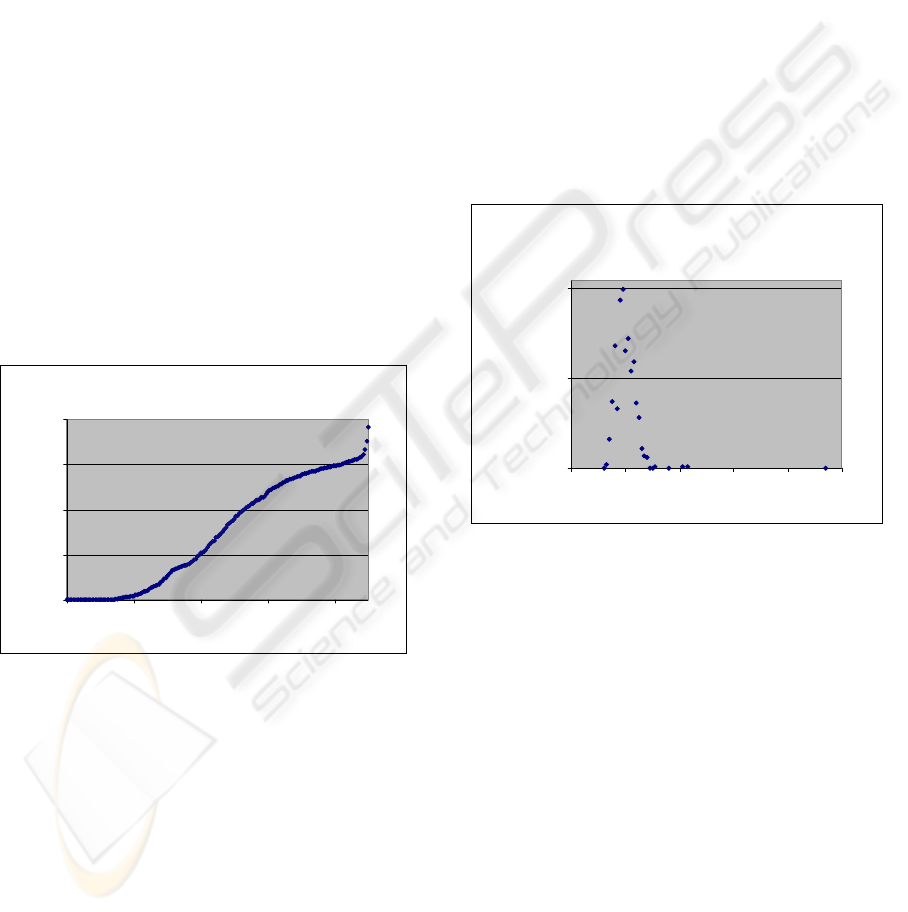
Figure 1: Each cluster should have at least one pair of
receptors shairing percentage of identity withen the TMD
above a certain threshold. Number of clusters converge to
one near 25% of identity. The horizontal axis shows the
sequence identity threshold while the vertical one shows
the number of clusters.
The process is continued until all pairs of receptors
are evaluated. Each receptor in each cluster should
share sequence identity less than x% with at least
one other receptor. The number of the clusters in
each threshold and the shape of the obtained graph
could be an index for the cumulative sequence
identity within the family or subfamily. If the
number of clusters converge to 1 in high threshold,
then we should conclude that the cumulative
sequence identity is high. Number of clusters
converge to one near 25% of identity in TMDs of
human GPCRs (figure 1) while it is in 42% and
37% of identity in amine and peptide subfamilies
respectively.
Similarity with Bovine Rhodopsin
Firstly, similarities within the TMDs were evaluated,
and then in order to evaluate the similarities in the
upper half (ligand binding domain) and the lower
half (G-protein binding domain) separately, each
helix was divided at its centre. The averaged
similarity of the whole TMDs was 19.7%. While in
the endoplasmic half of the TMDs, it was greater
than in the exoplasmic part. The average score for
the endoplasmic half of the rhodopsin-like hGPCRs
is 25.0% while for the exoplasmic half, it is 14.1%.
Identity percentage to rhodopsin withen
TMDs
1
51
101
0 20406080100
Identiry percentage
Receptors count
Figure 2: Pair-wise alignment of each family A receptor in
the human genome with rhodopsin separately (only
TMDs). Only six receptors are above 30% of identity and
according to the well-known rules in the field of
homology modeling, X-ray structure of the TMDs of
rhodopsin could be employed for constructing models
with enough confidence.
From pair-wise alignment of all hGPCRs with
bovine rhodopsin, we obtained only six receptors
with sequence identity above a threshold of 30%.
And as depicted in figure 2, most of the receptors
have sequence identity around 20%. The need for a
detailed analysis of the similarity to bovine
rhodopsin stems from the question of usefulness of
the rhodopsin model as a template for constructing
other GPCRs. Any model construction must relate to
sequential parts of the structure and not to individual
positions in space. Therefore, it is important to
record the change in the similarity along each one of
the helices and to realize which parts may be
BIOSIGNALS 2009 - International Conference on Bio-inspired Systems and Signal Processing
294
considered to be “stable” enough so that a variation
of sequence will not affect their structures. The
conservation of sequence stretches of different
length was calculated. Each stretch begins from N to
C.
In this study, we employed a conservation
scoring of segments in order to examine the extent
of the single known GPCR structure of bovine
rhodopsin which should probably not be “copied” in
modeling of other GPCRs. It was shown previously
that most of the conservation takes place in the
endoplasmic parts of the TMD, but quantitative
evaluations were limited to the conservations of
single residues. In our study, we focused on
cumulative conservation, because structural
templates can not be constructed of isolated residues
that are disconnected. By computing the similarity
along stretches of residues, thus constructing a
“cumulative similarity”, we demonstrated the
quantitative aspects of the differences in
conservation between the more conserved
endoplasmic regions of most TM helices in
rhodopsin-like hGPCRs and the exoplasmic parts.
This has been attributed to the more prominent
structural roles of the endoplasmic parts, or to their
very similar function, to transmit a signal to
intracellular G-proteins. The high variability of the
exoplasmic parts probably reflect the need to interact
and to be specific to a wide range of ligands.
There are certainly other possibilities for
dividing the lengths of the transmembrane helices,
and these may be useful for further refinement. We
have shown that it is possible to determine the exact
number of residues in a “stretch” whose averaged
similarity to bovine rhodopsin does not exceed a
certain threshold. We have also employed the
“windows” method and found that then we could
have better chances to model hGPCRs based on
bovine rhodopsin than employing the whole set of
residues in the endoplasmic half (see figure 3a-3g).
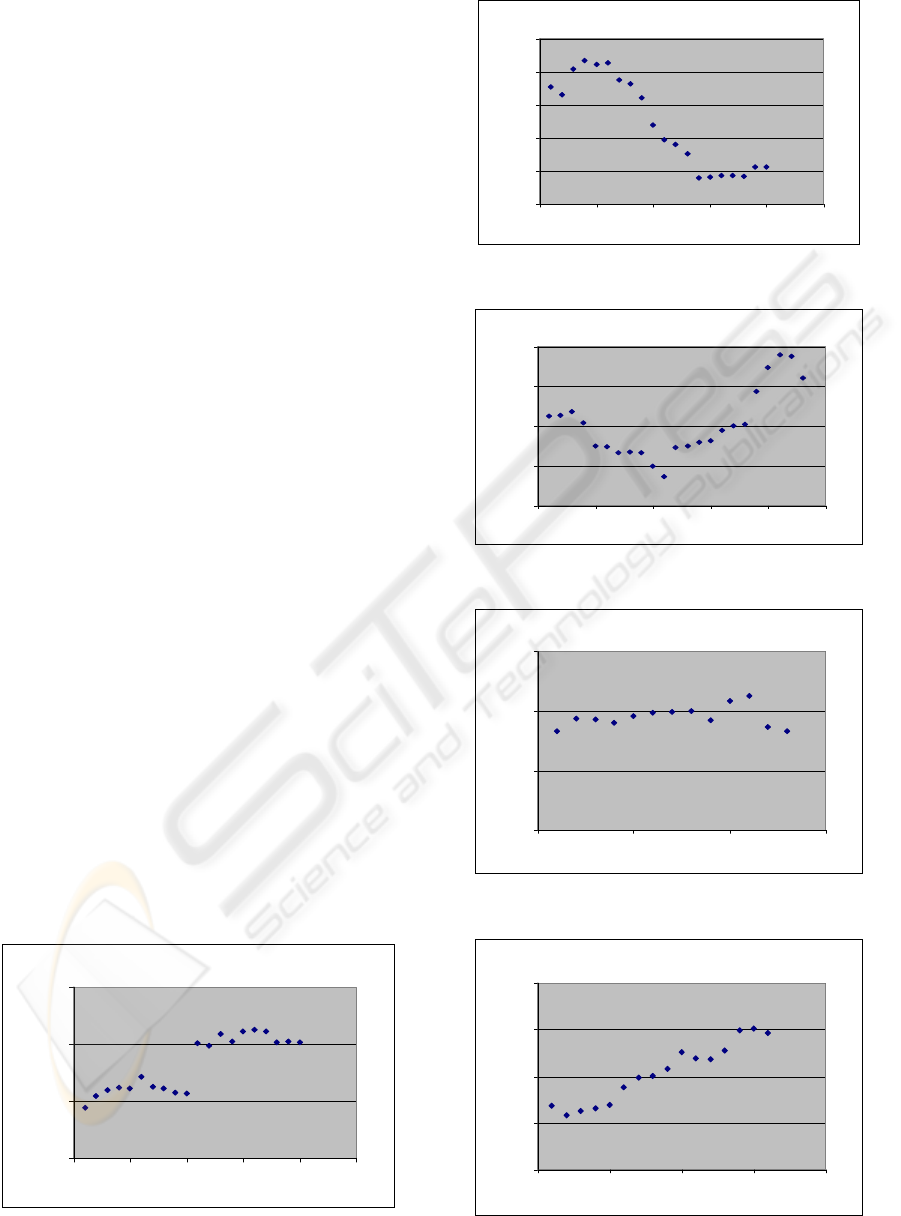
TM-1
0
10
20
30
0 5 10 15 20 25
First amino acid number
Averaged SI to Rh
Figure 3a
TM-2
0
10
20
30
40
50
0 5 10 15 20 25
First amino acid number
Averaged SI to Rh
Figure 3b
TM-3
0
10
20
30
40
0 5 10 15 20 25
First amino acid number
Averaged SI to Rh
Figure 3c
TM-4
0
10
20
30
0 5 10 15
First amino acid number
Averaged SI to Rh
Figure 3d
TM-5
0
10
20
30
40
0 5 10 15 20
First amino acid number
Averaged SI to Rh
Figure 3e
HOW MUCH BOVINE RHODOPSIN CRYSTAL STRUCTURE IS USEFUL FOR MODELING HUMAN GPCRS? -
ß2-Adrenergic Receptor as a Test Case
295
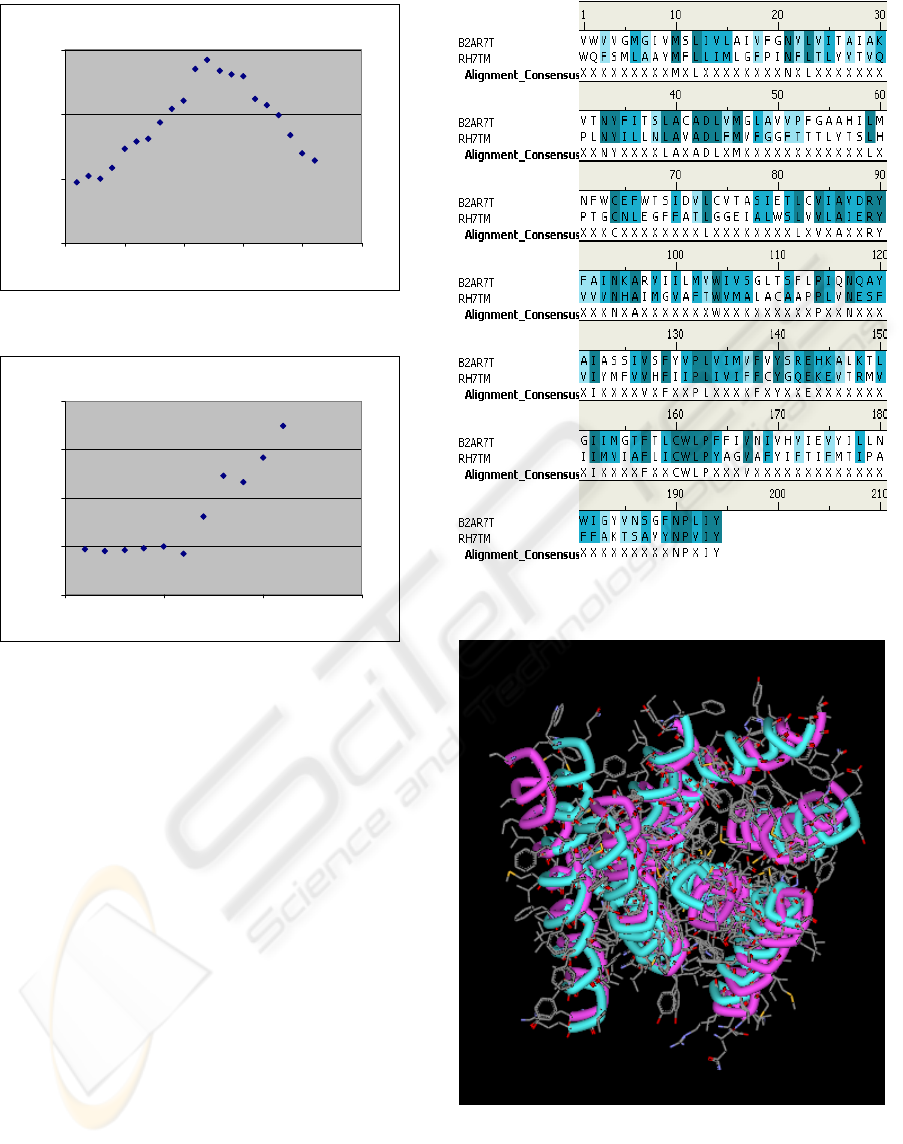
TM-6
0
10
20
30
0 5 10 15 20 25
First amino acid number
Averaged SI to Rh
Figure 3f
TM-7
0
10
20
30
40
0 5 10 15
First amino acid number
Averaged SI to Rh
Figure 3g
Figure 3a-3g: Averaged identity scores to bovine
rhodopsin (equation 1) of the seven TMDs of all family A
hGPCRs averaged over window of 11 residues. Horizontal
axis presents initial window positions. Y-axis is partial
conservation. The direction in each helix goes from the N-
terminal side to the C-terminal side.
Modeling of β2-Adrenergic Receptor based
on Bovine Rhodopsin as a Test Case
Since X-ray structure of β2-adrenergic receptor was
released recently, we have used it to validate our
findings that were obtained in this bioinformatics
study. In figure 4 we find the pairwise sequence
alignment of the transmembranal domains of β2-
adrenergic receptor and Bovine Rodopsin, while in
figure 5, the structural alignment is presented. The
best core segments that were selected according to
the findings as depicted in figure 3 gives backbone
RMSD equal 1.39 Å (see figure 6).
Figure 4: Pair-wise alignment of TMD of bovine
rhodopsin with β2-adrenergic receptor.
Figure 5: Superposition of TMD 3D structures, β2-
adrenergic receptor (2RH1) with Bovine Rodopsin (1F88).
The backbone RMSD is equal 2.05 Å. In general, the
upper half is more deviated than the lower half (mainly the
first three turns of transmembrane-1, left side shown in the
picture above).
BIOSIGNALS 2009 - International Conference on Bio-inspired Systems and Signal Processing
296

Figure 6: Superposition of the 7-transmembranal segments
composed of 11 residues each, that were selected based on
the bioinformatics analysis, β2-adrenergic receptor
(2RH1) with Bovine Rodopsin (1F88). The backbone
RMSD is equal 1.39 Å.
The shift in the cytoplasmic end of TMD-6 that
has been seen in the crystal structure of β2-
adrenergic receptor (Rasmussen et al., 2007) could
be explained by graph 3f. The segment of TMD-6 to
be used for modeling β2-adrenergic receptor based
on bovine rhodopsin in lying on the middle of the
helix.
Pair-wise alignment of the TMDs of family A
hGPCRs with β2-adrenergic receptor is shown in
figure 7. 103 receptors are above 30% of identity
and many others with identity less than 20%. We
will further test if we could obtain better models
while combining segments from the two crystal
structures (bovine rhodopsin and β2-adrenergic
receptor).
Identity Percentage to beta(2) adrenergic within
TMDs
1
21
41
61
81
101
121
020406080100
Ide ntity perce ntage
Receptors counts
Figure 7: Pair-wise alignment of each family A receptor in
the human genome with β2-adrenergic receptor separately
(only TMDs). 103 receptors are above 30% of identity.
4 CONCLUSIONS
We present in this study a qualitative and a
quantitative analysis of family A hGPCRs database
and tested the usefulness of employing crystal
structure of bovine rhodopsin as a template for
modeling the TMDs of other receptors from the
same family. In most cases, as shown in figures 3a-
3g, helix terminals display a smaller conservation
than other parts of the helices. This is also found in
most of the endoplasmic, more conserved parts
(except for helix VI that has a larger conservation
value at the middle). These variations could be
connected to the structural changes from helix to
loop at both the endoplasmic and exoplasmic
terminals. Structural analysis of the recently released
structure of β2-adrenergic receptor and superposition
of certain parts from the transmembranal domains
with Bovine Rhodopsin backed our findings that
were obtained by this study.
Since we are using only a partial template from
the TM helical region of bovine rhodopsin or β2-
adrenergic receptor, there still persists an immense
problem of determining the rest of the helical
coordinates. Based on the information extracted
from this study, we plan to use Molecular Dynamics
(MD), Simulated Annealing (SA) or Iterative
Stochastic Elimination (ISE)
(http://www.pdb.org/pdb/explore.do?structureId=1F
88) in order to construct better models for GPCRs,
starting with a partial template of rhodopsin and/or
β2-adrenergic receptor.
ACKNOWLEDGEMENTS
We gratefully acknowledge RAND Biotechnologies
ltd company for providing us with the database of
rhodopsin like hGPCRs and the Trans-membrane
domains allocations module.
REFERENCES
Gether U., (2000). Uncovering molecular mechanisms
involved in activation of G Protein-Coupled
Receptors. Endocr Rev, 21, 90-113
Nurnberg B., Gudermann T., Schultz G., (1995).
Receptors and G proteins as primary components of
transmembrane signal transduction. J Mol Med, 73,
123-132
Drews J., (2000). Drug discovery: a historical perspective.
Science, 287, 1960-1964
HOW MUCH BOVINE RHODOPSIN CRYSTAL STRUCTURE IS USEFUL FOR MODELING HUMAN GPCRS? -
ß2-Adrenergic Receptor as a Test Case
297

Nambi P. & Aiyar N., (2003). G protein-coupled receptors
in drug discovery. Assay and Drug Development
Technologies, 1, 305-310
Bissantz C., Bernard P., Hibert M. & Rognan D., (2003).
Protein-based virtual screening of databases. II. Are
homology modeling of G-protein coupled receptors
suitable targets. Proteins – Structure Function and
Genetics, 50, 5-25
Palczewski K., Kumasaka T., Hori T., Behnke C.A.,
Motoshima H., Fox B.A. Le Trong I., Okada T.,
Stenkamp R.E., Yamamoto M. & Miyano M. (2000).
Crystal structure of rhodopsin: a G-protein coupled
receptor. Science, 289(5480), 739-45
Okada T., Nakamichi H., (2004). X-ray crystallography of
rhodopsin. Phase Transitions, 77, 21-29
Rasmussen S.G.F., Choi H.-J., Rosenbaum D.M., Kobilka
T.S., Thian F.S., Edwards P.C., Burghammer M.,
Ratnala V.R.P., Sanishvili R., Fischetti R.F., Schertler
G.F.X., Weis W.I. & Kobilka B.K. (2007). Crystal
structure of the human β
2
adrenergic G-protein-
coupled receptor. Nature, 450(7168), 383-387
Cherezov V., Rosenbaum D.M., Hanson M.A., Rasmussen
S.G.F., Thian F.S., Kobilka T.S., Choi H.-J., Kuhn P.,
Weis W.I., Kobilka B.K. & Stevens R.C. (2007).
Science, 23, 318(5854), 1258-1265
Rayan A., Siew N., Cheno Schwartzs S., Matzner Y.,
Bautsch W. Goldblum A., (2000), A novel
computational method for predicting the
transmembranal structure of G-protein coupled
receptors: application to the human C5aR and C3aR.
Receptors Channels, 7(2), 121-137
Eszter H., Zsolt B., (2008), Homology modeling of breast
cancer resistance protein (ABCG2). Journal of
Structural Biology, 162, 63-74
http://www.gpcr.org/7tm/ (June 2006 release (10.0))
Oliveira L., Paiva A.C.M., Vriend G., (2002), Correlated
mutation analyses on very large sequence families.
Chembiochem, 3, 1010-1017
Shacham S., et al. (2001). Modeling the 3D structure of
GPCRs from sequence. Med Res Rev, 21, 472-483
Baker D., Sali A., (2001). Protein structure prediction and
structural genomics. Science 294 (5540), 93-96
Baldwin J.M., Schertler G.F., Unger V.M. (1997). An
alpha-carbon template for the transmembrane helices
in the rhodopsin family of G-protein-coupled
receptors. J Mol Biol, 272, 144-164
Mirzadegan T., Benko G., Filipek S., Palczewski K.,
(2003). Sequence analyses of G-protein-coupled
receptors: Similarities to rhodopsin. Biochemistry, 42,
2759-2767
Rayan A.M., Raiyn J.A., 2008. Intelligent Learning
Engine (ILE) Optimization Technology, provisional
patent
TMDs
-Scanner software
http://www.pdb.org/pdb/explore.do?structureId=1F88
Rayan A., Noy E., Chema D., Levitzki A., Goldblum A.,
2004. Stochastic algorithm for kinase homology model
construction. Current Medicinal Chemistry, 11, 675-
692
BIOSIGNALS 2009 - International Conference on Bio-inspired Systems and Signal Processing
298
