
STATISTICAL MECHANICS OF PROTEINS IN THE RANDOM
COIL STATE
Cigdem Sevim Bayrak and Burak Erman
Computational Science and Engineering Program, Koc University, 34450, Sariyer, Istanbul, Turkey
Keywords: Rotational isomeric state, Random coil, Denatured state.
Abstract: Denatured proteins are mostly partially folded and compact proteins. A statistical analysis on
thermodynamic properties is presented to describe and characterize denatured proteins. Conformational free
energy, energy, entropy and heat capacity expressions are derived using the Rotational Isomeric States
model of polymer theory. The state space and the probabilities of each state are comprised from a coil
database. Properties for the denatured state are obtained for a sample set of proteins taken from the Protein
Data Bank. Thermodynamic expressions of denatured state are derived.
1 INTRODUCTION
Random configurations of protein chains are
obtained under the constraints imposed by chain
connectivity and the torsion states of the backbone
torsion angles
and
in the absence of sequence-
distant long-range interactions. The term ‘randomly
coiled proteins’ describing this state have been
studied in detail by Flory and collaborators, based on
the Rotational Isomeric States (RIS) Model of
polymer theory (Flory, 1969); (Brant and Flory,
1965); (Brant et al., 1969); (Conrad and Flory,
1976); (Flory and Jernigan, 1965); (Rehahn et al.,
1997); (Engin et al., 2009).The RIS model for a
protein chain consists of two major components: (1)
The statistical weights of the torsion states of the
and
angles, and (2) The proper matrix
multiplication operations leading to the partition
function of the chain. Thermodynamics of the single
chain then follows upon proper matrix operations
based on the partition function and its derivatives
(Callen, 1985); (Flory, 1974). Understanding the
random configurations of proteins is important due
to several reasons: Firstly, the set of random
configurations covers all possible initial
conformations of proteins. Depending on the
primary sequence, some conformations emerge as
highly probable due to the amino acid specific
regions of the
(, )
angles. Secondly, under
strongly denaturing conditions, a wide range of
values become available to
and
, and
conformations are close to those of the random coil
(Dill and Shortle, 1991); (Tanford, 1968). These
conformations are many in number, and therefore a
statistical characterization is required to understand
the thermodynamics of the denatured state. Thirdly,
the functionally important ‘intrinsically disordered
protein’ concept where the primary sequence
prohibits the folded state, may suitably be analyzed
by the tools used to understand the random
conformations (Tompa, 2011); (Orosz and Ovádi,
2011).
Thus, a better statistical understanding of
denatured proteins is required for answering
questions referring to functional properties of
proteins. The number of states available to the
denatured chain may vary from an enormous set to
only a few in numbers as observed in switches. The
general statistical mechanical model that we adopt is
not restricted with this variation. The size of
available states is determined by the probabilities of
the latter, and several sources for such probabilities
are either available and may be extracted from
various databases, or may be generated by suitable
training techniques of bioinformatics, depending on
the constraints and requirements of the problem at
hand. In the present study, we extract the
probabilities from the Ramachandran plots obtained
from the coil library (Fitzkee et al., 2005) which is
accepted to be representative of the random coiled
state of proteins (Ormeci et al., 2007); (Engin et al.,
2009); (Unal et al., 2010). Having characterized the
probabilities from the knowledge data base, we
220
Sevim Bayrak C. and Erman B..
STATISTICAL MECHANICS OF PROTEINS IN THE RANDOM COIL STATE.
DOI: 10.5220/0003785202200225
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2012), pages 220-225
ISBN: 978-989-8425-90-4
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

apply the matrix multiplication technique to obtain
the partition function, and the thermodynamic
functions such as energy, entropy and heat capacity
for the denatured state. Finally we present random
coil results for thermodynamic functions for several
proteins whose primary sequences are chosen from
the Protein Data Bank.
2 STATISTICAL EVALUATION
A denatured protein assumes a multitude of
conformations, each subject to a certain probability
determined by the configurational features of the
residues which are either of local or nonlocal nature.
Local effects result from interactions among
neighboring amino acids along the chain. We refer
to this state the random coiled state of the protein.
Determination of the conformation of a chain using
near neighbor interactions only reduces the problem
to a Markov process. Nonlocal effects are those
among residues separated by more than two residues
along the chain. Having adopted the probabilities
from the coil library, where the sequence-distant
long-range interaction are absent because secondary
or tertiary structures are lacking, is a good
approximation to the Markov nature of the coiled
state.
Markov statistics of denatured proteins have an
important place in protein statistics in general,
because: (i) This is the first approximation to the
difficult problem of non-Markov behavior, (ii)
Markov behavior is responsible for a large body of
observed phenomena, (iii) There is already a
powerful and successful Markov model of
characterizing the conformations of polymers, i.e.,
the Rotational Isomeric States (RIS) model that has
been studied in some detail. The specific aim of the
present paper is to extend the RIS model to calculate
the thermodynamic properties of denatured chains
using data generated from the denatured components
of chains from the PDB.
Rotational Isomeric State (RIS) formalism
(Flory, 1969) replaces the continuous distribution of
backbone torsion angles by a distribution over
several discrete states, and integrals over the energy
surface are approximated by summations over these
states. The native state of a protein is obtained when
each torsion angle selects a single unique value. Two
torsion angles around the alpha carbon,
C
,
describe the local conformation of a residue. The
Flory isolated pair hypothesis suggests that each pair
of torsion angles is independent of the angles
occupied by neighboring pairs (Brant and Flory,
1965); (Flory, 1969). Rose and coworkers. Zaman et
al., (2003), Jha et al., (2005) and Keskin et al.,
(2004), Esposito et al., (2005) and Colubri et al.,
(2006) showed the existence of significant
correlations between neighboring torsion angle
pairs. In a recent work it has been shown that the
usage of
1
,
ii
provides more information on
backbone behavior as opposed to independent usage
of residues (Lennox et al., 2009).
Some values of torsion angles are more favorable
than others, and different amino acid types have
different propensities to occur in different angles
(Karplus, 1996). The dependence between the
torsion states of two neighboring residues is a
function of the type of the residues (Keskin et al.,
2004). We elaborate further on this point in
discussing the construction of energy maps below.
Figure 1: Torsion angles of the i
th
amino acid.
The frequency of occurrence of a given amino
acid at a given torsion state leads to the probabilities.
For calculations of the random denatured
conformations of proteins, a coil library serves as the
source of information where torsion angle data is
taken from the set of amino acids those are not in
helical or beta structures. In this paper, we use the
Rose Protein Coil Library (Fitzkee et al., 2005).
2.1 States
The backbone torsion angles for the ith amino acid
are shown in Figure 1. Each bond can assume
different angles, with different preferences. Each
residue has three torsion angles,
, , and
. The
occurrence of a residue in a given
and
state,
irrespective of its type is presented in Figure 1. An
examination of this figure shows that the choice of
isomeric states for the
and
angles is more
complicated than the choice in synthetic polymer
applications. In the latter, usually there are a few
states like trans, gauche+ and gauche-, and their
combinations for two successive bonds along the
chain. In the protein case, there are several discrete
states centered on different regions for the
successive
and
angles, and for different amino
acids.
STATISTICAL MECHANICS OF PROTEINS IN THE RANDOM COIL STATE
221

We construct state probabilities over the
Ramachandran map for each residue. 13 states are
identified for the following
axis intervals: (-180,-
150), (-150,-120), (-120,-105), (-105,-75), (-75,-40),
(-40,-20), (-20,-10), (-10,30), (30,70), (70,105),
(105,130), (130,155), (155,180). The corresponding
intervals over
axis are: (-180,-160), (-160,-135),
(-135,-105), (-105,-75), (-75,-40), (-40,-15), (-15,
20), (20, 60), (60, 90), (90,110), (110,130),
(130,160). For the
angle, there are two states,
one is either (-180,-160) or (160,180), and the other
is (-20, 20). The states chosen in this manner are
representative of the regions given by Karplus
(1996) and also in (Unal et al., 2009). Thus, we
identified 13 states for the angle
, 13 states for
,
and 2 states for
as rotational isomeric states.
2.2 State Probabilities
The pair wise dependent probabilities of observed
states of angles are defined as
11
,,
,,
,,
Xii Xii X
Xii Xii X
X
Yii XYii XY
PN N
PN N
PN N
(1)
where
,
Xii
N
is the number of residue type X
observed in the indicated states, and
X
N
is the
total number of conformations (Keskin et al., 2004);
(Unal et al., 2009). Similarly,
1
,
XY i i
N
is the
number of dipeptides of XY in the given
conformations. Here,
,
Xii
P
and
,
Xii
P
are the probabilities of observing residue X to be in
state
,
ii
, and in state
,
ii
respectively.
1
,
XY i i
P
is the joint probability of observing
residue X in state
i
and Y in state
1i
. The
neighbor-dependence introduced in the third of (1
) is
a dependence that originates from the residue type
differences. Otherwise, (1
) acknowledge the Flory
isolated pair hypothesis. The conformational
energies are defined as
00
00
1
1
00
1
,
,ln
,
,ln
,
,ln
Xii
Xii
XiX i
Xii
Xii
XiXi
XY i i
XY i i
XY i XY i
P
ERT
PP
P
ERT
PP
P
ERT
PP
(2)
where the superscript 0 indicates the uniform
distribution probabilities. Hence, they are directly
proportional to the size of the angular intervals;
00
113
Xi X i
PP
and
0
12
Xi
P
.
Statistical weights
ii
u
,
ii
u
, and
1ii
u
corresponding to the energies may be defined by
1
;
;
;1
exp , /
exp , /
exp , /
ii
ii
ii
XXii
XXii
XY XY i i
uERT
uERT
uERT
(3)
where R is the gas constant, T is the temperature.
The statistical weight matrix for a configuration
can be written as a product of statistical weights of
each bond pair,
,, ,, and ,
. For this
purpose, the statistical weight matrix for a given
residue X is defined as
ii
X
X
Uu
,
ii
X
X
Uu
, and
1ii
XY
XY
Uu
. Depending
on the number of states of each angle, dimensions of
the statistical weight matrices
X
U
,
X
U
, and
XY
U
, are 13×13, 13×2, and 2×13, respectively. The
superscripts
,
,
,
, and
,
identify the
bond pairs over which statistical weights are
calculated.
2.3 Calculation of the Thermodynamic
Quantities
The partition sum of statistical weights for all
configurations of the chain is given by (Flory, 1974)
*
2
1112
nn
Z
JUUUUU UU J
(4)
where
*
10 0J
,
11 1J column
.
The thermodynamic properties, and the
coefficients derived from them depend not only on a
single conformation of the peptide, but on all
possible configurations. In the remaining equations,
we give the relevant expressions for calculating
these averages.
2.3.1 Helmholtz Free Energy
Since the Helmholtz free energy in canonical
formalism is additive over the energies, it can be
calculated using the partition function of the chain
(Callen, 1985).
ln
F
Z
(5)
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
222

where, 1/kT
.
2.3.2 Mean Energy
The average energy is given by
1
ln
ddZ
EZ
dZd
(6)
The matrix multiplication formalism of the partition
function leads to matrix multiplication scheme of its
derivatives in the following way
*
ˆ
i
dZ
L
UL
d
(7)
where
**
00LJ
,
00L column J
and Û is the super matrix whose elements are
matrices
ˆ
0
UU
U
U
(8)
dU
U
d
(9)
Therefore, the mean energy can be obtained using
the following multiplication scheme
*
1
i
E
LGL
Z
(10)
where
0
i
i
UU
G
U
(11)
2.3.3 Entropy
The entropy of the chain can be expressed in terms
of Z and its derivatives with respect to β. Following
the equality
2
Sk dFd
, is obtained.
2
2
11ln
ln ln
SZ
Z
ZE
k
(12)
Using the matrix multiplication formalism of Z and
its first derivative with respect to
, the entropy can
be calculated as
*
*
*
ln
i
i
i
L
GL
S
JUJ
k
J
UJ
(13)
2.3.4 Heat Capacity
The heat capacity is one of the most important
properties of the proteins, both native and denatured.
When force acting on the chain is taken as zero,
denoted below by the subscript
0f , the heat
capacity can be calculated as
2
2
0
2
0
ln
f
f
E
Z
Ck
T
(14)
Similar to (7), second derivative can be obtained as
2
*
2
ˆ
ˆ
i
Z
M
UM
(15)
where
**
000000MJ
and
000000
M
column J
, and
00
ˆ
ˆ
00
00 0
UUUU
UU
U
UU
U
(16)
2
'''
2
,
dU U
UU
d
(17)
The second derivative of
ln
Z
on the right hand side
of the equation is written in terms of the first and
second derivatives of the partition function:
2
22
22 2
ln 1 1
Z
ZZ
Z
Z
(18)
Hence the heat capacity to be calculated by the
matrix notation
0
2
*
*
2
**
ˆ
ˆ
ˆ
f
i
i
ii
MUM
LUL
Ck
JUJ JUJ
(19)
3 RESULTS
In this section, the free energy, energy, entropy, and
heat capacity of peptides of different sizes ranging
from 10 to 800 amino acids are calculated using the
RIS model, over a temperature range of 200-700 K.
Table 1 lists the protein set taken from the PDB.
The variation of the free energy, energy, entropy
and heat capacity is evaluated by repeating the
calculations. Results are presented in Figure 2.
The curves shown in the four panels of Figure 2
are not independent from each other, and are related
by the thermodynamic relations given by (5), (6),
(12), and (14). It is seen that the curves in the figures
all scale with the number of residues N.
STATISTICAL MECHANICS OF PROTEINS IN THE RANDOM COIL STATE
223
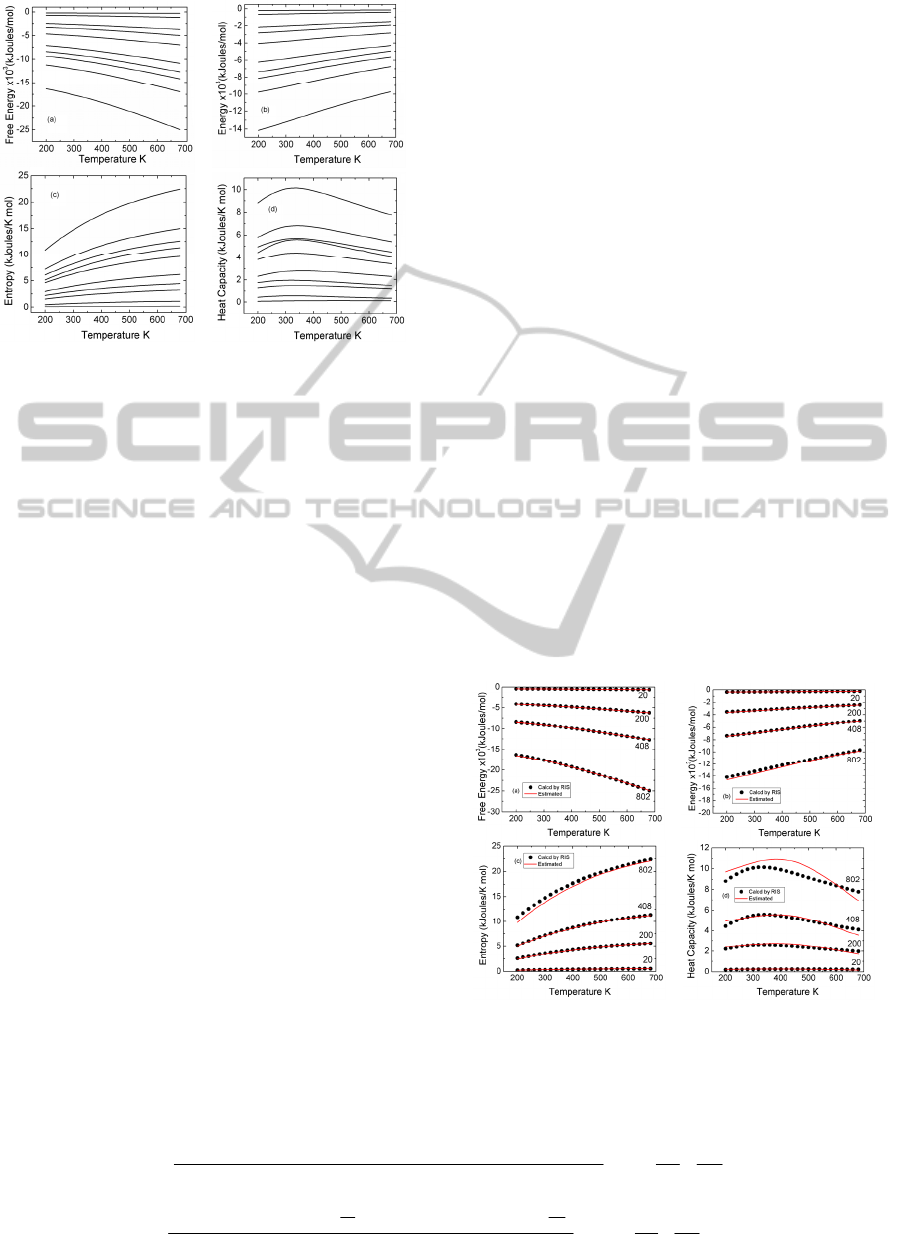
Figure 2: (a) The free energy as a function of temperature,
T, for different length proteins. (b) energy as a function of
T. (c) entropy as a function of T. (d) heat capacity as a
function of T. The curves in parts (a),(b), and (c) are
ordered from top to bottom represent proteins with the
following numbers of residues: 10, 40, 120, 160, 226, 349,
408, 456, 545, and 802, respectively. In part (d) they are in
reverse order.
In order to find analytical functions that will give
the curves shown in Figure 2, we first chose an
analytical form for the heat capacity as
3
0
(, )
BT DT
f
CTNNTAe Ce
(20)
keeping in mind the thermodynamic postulates. We
inspired the Debye model of heat capacity in a solid
that shows the dependence of T
3
. Then, by
integration subject to the conditions imposed by (5),
(6), (12), and (14), we obtain the remaining
thermodynamic functions as given in Eqs. (21)-(23).
We obtain the coefficients of (20)-(23) by curve
fitting as
6
1.5 10A
kJoules/K
4
mol,
3
7.2 10B
1/K,
5
2.6 10C
kJoules/K
4
mol,
2
2.3 10D
1/K, and 4083E kJoules/mol.
4 CONCLUSIONS
The use of the RIS model depends critically on two
items: (i) the choice of the states, and (ii) the choice
of the database with which the probabilities of these
states are evaluated. The states are described in
terms of the populated regions on the Ramachandran
map, and the possible states for the
and
angles
of different amino acids are determined following
the work of Karplus (Karplus, 1996). In order to
apply the RIS model, however, the states available
to the torsion angles
, , and
are required
separately. The state space is obtained in our
formulation as 13 states for
and 13 states for
,
and two states for
. Evaluation of the probabilities
follows the choice of the state space. For proof of
principle, we used a coil library for the
determination of the probabilities. One could
alternatively construct a databank of known
denatured proteins, or a subset of them depending on
the nature of the investigation. Once the states are
determined, the RIS model is independent of the
databases used. We observed that the per residue
thermodynamic properties of proteins in the random
coil state scales only with the temperature. While
entropy and energy increases with the temperature,
free energy decreases. Heat capacity represents a
decrease around 340 Kelvin that implies an energy
barrier for a possible transition state. The explicit
expressions that we determined for the
thermodynamic functions form a thermodynamically
consistent set which may be used to obtain other
thermodynamic potentials by applying the known
Legendre transformation techniques (Callen, 1985).
Figure 3: Comparison of (a) free energy, (b) mean energy,
(c) entropy, and (d) heat capacity estimates. Exact values
are calculated by matrix multiplication scheme, estimated
values are calculated by fundamental relation. The lengths
of chains are shown on each curve.
322 3 22
33 3 3
( 2 2) ( 2 2)
(, ) 2
BT DT
A
DNeBTBT CBNeDTDT AC
ST N N
BD B D
(21)
322 3 22
33 3 3
66
(4) ( 4)
(, ) 2
BT DT
AD Ne B T T CB Ne D T T
AC
BD
FT N NT EN
BD B D
(22)
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
224

3232 3 232
33
66
(36) ( 36)
BT DT
AD Ne B T BT T CB Ne D T DT T
BD
UFTS EN
BD
(23)
REFERENCES
Brant, D. A. and Flory, P. J. 1965. The Configuration Of
Random Polypeptide Chains. Ii. Theory. Journal Of
The American Chemical Society, 87, 2791-2800.
Brant, D. A., Tonelli, A. E. & Flory, P. J. 1969. The
Configurational Statistics Of Random Poly(Lactic
Acid) Chains. Ii. Theory. Macromolecules, 2, 228-
235.
Callen, H. B. 1985. Thermodynamics And An Introduction
To Thermostatistics, New York, Wiley.
Colubri, A., Jha, A. K., Shen, M. Y., Sali, A., Berry, R. S.,
Sosnick, T. R. & Freed, K. F. 2006. Minimalist
Representations And The Importance Of Nearest
Neighbor Effects In Protein Folding Simulations. J
Mol Biol, 363, 835-57.
Conrad, J. C. & Flory, P. J. 1976. Moments And
Distribution Functions For Polypeptide Chains. Poly-
L-Alanine. Macromolecules, 9, 41-47.
Dill, K. A. & Shortle, D. 1991. Denatured States Of
Proteins. Annu Rev Biochem, 60, 795-825.
Engin, O., Sayar, M. & Erman, B. 2009. The Introduction
Of Hydrogen Bond And Hydrophobicity Effects Into
The Rotational Isomeric States Model For
Conformational Analysis Of Unfolded Peptides. Phys
Biol, 6, 016001.
Esposito, L., De Simone, A., Zagari, A. & Vitagliano, L.
2005. Correlation Between [Omega] And [Psi]
Dihedral Angles In Protein Structures. Journal Of
Molecular Biology, 347, 483-487.
Fitzkee, N. C., Fleming, P. J. & Rose, G. D. 2005. The
Protein Coil Library: A Structural Database Of
Nonhelix, Nonstrand Fragments Derived From The
Pdb. Proteins, 58, 852-4.
Flory, P. J. 1969. Statistical Mechanics Of Chain
Molecules, New York,, Interscience Publishers.
Flory, P. J. 1974. Foundations Of Rotational Isomeric
State Theory And General Methods For Generating
Configurational Averages. Macromolecules, 7, 381-
392.
Flory, P. J. & Jernigan, R. L. 1965. Second And Fourth
Moments Of Chain Molecules, Aip.
Jha, A. K., Colubri, A., Freed, K. F. & Sosnick, T. R.
2005. Statistical Coil Model Of The Unfolded State:
Resolving The Reconciliation Problem. Proc Natl
Acad Sci U S A, 102, 13099-104.
Karplus, P. A. 1996. Experimentally Observed
Conformation-Dependent Geometry And Hidden
Strain In Proteins. Protein Sci,
5, 1406-20.
Keskin, O., Yuret, D., Gursoy, A., Turkay, M. & Erman,
B. 2004. Relationships Between Amino Acid
Sequence And Backbone Torsion Angle Preferences.
Proteins: Structure, Function, And Bioinformatics, 55,
992-998.
Lennox, K. P., Dahl, D. B., Vannucci, M. & Tsai, J. W.
2009. Density Estimation For Protein Conformation
Angles Using A Bivariate Von Mises Distribution And
Bayesian Nonparametrics. J Am Stat Assoc, 104, 586-
596.
Ormeci, L., Gursoy, A., Tunca, G. & Erman, B. 2007.
Computational Basis Of Knowledge-Based
Conformational Probabilities Derived From Local-
And Long-Range Interactions In Proteins. Proteins:
Structure, Function, And Bioinformatics, 66, 29-40.
Orosz, F. & Ovádi, J. 2011. Proteins Without 3d Structure:
Definition, Detection And Beyond. Bioinformatics, 27,
1449-1454.
Pappu, R. V., Srinivasan, R. & Rose, G. D. 2000. The
Flory Isolated-Pair Hypothesis Is Not Valid For
Polypeptide Chains: Implications For Protein Folding.
Proc Natl Acad Sci U S A, 97, 12565-70.
Rehahn, M., Mattice, W. L. & Suter, U. 1997. Rotational
Isomeric State Models In Macromolecular Systems,
Springer.
Tanford, C. 1968. Adv. Protein Chem., 23, 121-282.
Tompa, P. 2011. Unstructural Biology Coming Of Age.
Curr Opin Struct Biol, 21, 419-25.
Unal, E. B., Gursoy, A. & Erman, B. 2009.
Conformational Energies And Entropies Of Peptides,
And The Peptide-Protein Binding Problem. Physical
Biology, 6.
Unal, E. B., Gursoy, A. & Erman, B. 2010. Vital: Viterbi
Algorithm For De Novo Peptide Design. Plos One, 5,
E10926.
Zaman, M. H., Shen, M.-Y., Berry, R. S., Freed, K. F. &
Sosnick, T. R. 2003. Investigations Into Sequence And
Conformational Dependence Of Backbone Entropy,
Inter-Basin Dynamics And The Flory Isolated-Pair
Hypothesis For Peptides. Journal Of Molecular
Biology, 331, 693-711.
STATISTICAL MECHANICS OF PROTEINS IN THE RANDOM COIL STATE
225
