
Homozygosity Mapping using Whole-Exome Sequencing: A Valuable
Approach for Pathogenic Variant Identification in Genetic Diseases
Jorge Oliveira
1,2,*
, Rute Pereira
1,*
, Rosário Santos
2
and Mário Sousa
1
1
Instituto de Ciências Biomédicas Abel Salazar (ICBAS), Universidade do Porto,
R. Jorge de Viterbo Ferreira nº 228, Porto, Portugal
2
Centro de Genética Médica Dr. Jacinto Magalhães, Centro Hospitalar do Porto,
Praça Pedro Nunes nº88, Porto, Portugal
Keywords: Whole-Exome Sequencing, Homozygosity Mapping, Next-Generation Sequencing, Clinical Genetics.
Abstract: In the human genome, there are homozygous regions presenting as sizeable stretches, or ‘runs’ of
homozygosity (ROH). The length of these ROH is dependent on the degree of shared parental ancestry,
being longer in individuals descending from consanguineous marriages or those from isolated populations.
Homozygosity mapping is a powerful tool in clinical genetics. It relies on the assumption that, due to
identity-by-descent, individuals affected by a recessive disease are likely to have homozygous markers
surrounding the disease locus. Consequently, the analysis of ROH shared by affected individuals in the
same kindred often helps to identify the disease-causing gene. However, scanning the entire genome for
blocks of homozygosity, especially in sporadic cases, is not a straight-forward task. Whole-exome
sequencing (WES) has been shown to be an effective approach for finding pathogenic variants, particularly
in highly heterogeneous genetic diseases. Nevertheless, the huge amount of data, especially variants of
unknown clinical significance, and the presence of false-positives due to sequencing artifacts, makes WES
analysis complex. This paper briefly reviews the different algorithms and bioinformatics tools available for
ROH identification. We emphasize the importance of performing ROH analysis using WES data as an
effective way to improve diagnostic yield.
1 INTRODUCTION
Mendelian diseases are caused by pathogenic
variants in genes that follow the biological
inheritance laws originally proposed by Gregor
Mendel. The disease-causing gene may be in an
autosome or in a sex-chromosome, and may be
dominant or recessive. Individually, these diseases
are considered to be rare, but collectively they occur
at a high rate, with an estimated 7.9 million children
being born annually with a serious birth defect of
genetic origin (Christianson et al., 2006).
*
Sanger sequencing has been the gold standard in
molecular diagnostics of Mendelian diseases and is
still the first choice to confirm a suspected diagnosis,
enabling accurate genetic counselling. However, for
diseases with genetic heterogeneity, such as
hereditary myopathies or primary ciliary dyskinesia,
gene-by-gene Sanger sequencing is not the most
*
Equally contributing authors.
cost-effective or efficient approach (Oliveira et al.,
2015; Pereira et al., 2015). Technological advances
over the past decade has led to the development of
high-throughput sequencing platforms, revolutioni-
zing the sequencing capabilities and boosting the use
of this so called “next-generation sequencing”
(NGS) both in research and in clinical diagnostic
settings. With this technology, the human genome
can be completely sequenced, allowing the
simultaneous analysis of multiple genes. The
application of NGS in Mendelian diseases focuses
firstly on exonic regions of DNA, as the majority of
disease-causing mutations are found in exons or in
the flanking intronic regions (Ng et al., 2010).
Marriage between close biological relatives
increases the probability of the offspring inheriting
two deleterious copies of a recessive gene. Thus,
children from consanguineous couples have a higher
incidence of autosomal recessive disorders (Bittles,
2001). In addition, as alleles are parts of haplotypes,
not only will the affected descendant have two
identical copies of the ancestral allele, but the
210
Oliveira J., Pereira R., Santos R. and Sousa M.
Homozygosity Mapping using Whole-Exome Sequencing: A Valuable Approach for Pathogenic Variant Identification in Genetic Diseases.
DOI: 10.5220/0006248502100216
In Proceedings of the 10th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2017), pages 210-216
ISBN: 978-989-758-214-1
Copyright
c
2017 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

surrounding DNA segment (haplotype) will also be
homozygous. Thus, the child will be homozygous
for that segment, the so-called runs of homozygosity
(ROH) (McQuillan et al., 2008). These homozygous
segments that are identical by descent (IBD) are
generally longer in consanguinity cases.
Nevertheless, even in the absence of known recent
inbreeding, ROH can be detected in geographically
isolated populations and historical bottlenecks
(Pemberton et al., 2012). The ROH length is
dependent on the degree of shared parental ancestry
and its age. Recent inbreeding events/parental
consanguinity tend to have longer ROH (measuring
tens of Mb) since there are fewer recombination
events interrupting the segments that are IBD.
Conversely, older ROH are generally much shorter
because the homozygous stretches have been split
down by repeated meioses over the generations, with
the exception of genomic regions where the
recombinetion rates are lower (McQuillan et al.,
2008).
2 HOMOZYGOSITY MAPPING
Homozygosity mapping (also known as autozygosity
mapping), consists in the identification of
homozygous regions in the genome. This is a
powerful strategy
to associate new genes to diseases
(Alkuraya 2010; Goodship et al., 2000). As
mentioned, affected individuals are likely to have
two IBD alleles at markers located in the vicinity of
the disease locus and thus will be homozygous for
these markers. This method relies on the search for
ROH that are shared by affected individuals in the
same family. However, scanning the genome for
blocks of homozygosity, although simple and
efficient, requires sophisticated techniques such as
the use of numerous microsatellite markers or high-
density single nucleotide polymorphism (SNP)
genotyping. For most autozygosity mapping
projects, multipoint linkage analyses under a
recessive disease model have been performed, with
software such as GENEHUNTER (Kruglyak et al.,
1996), SIMWALK2 (Sobel et al., 2002), MERLIN
(Abecasis et al., 2002) or ALLEGRO (Gudbjartsson
et al., 2000).
In general, haplotypes are inspected manually for
homozygous regions that are shared by all affected
individuals, and can be inferred to be IBD if
genotypes from the parents or other close relatives
are available. But, in practical terms, conventional
parametric methods of multipoint linkage analysis
for large datasets in complex consanguineous
families are often difficult, because of the time and
computational power required, since homozygous
blocks among affected individuals tend to be large
(mean 4.4 Mb) and contain dozens or hundreds of
genes. Moreover, genotyping errors of SNP array
platforms and poorly represented chromosomal
regions also limit the potential of this technology. If
ROH are incorrectly mapped by the introduction of
erroneous heterozygous genotypes, the analysis of
the causative gene will be compromised.
3 NEXT-GENERATION
SEQUENCING
NGS, and in particular whole-exome sequencing
(WES), prompted great advances in the study of
genetic diseases and gene discovery (Boycott et al.
2013; Ng et al., 2010). However, this technology has
still some limitations (Sirmaci et al., 2012). Firstly,
some deleterious variations may be in non-coding
regions, which cannot be detected by WES. Genetic
and phenotypic heterogeneity in affected individuals
makes exome sequencing difficult to interpret.
Sequencing errors related to poor capture efficiency,
mechanical and analytical errors, as well as
misalignment of repetitive regions, lead to erroneous
results. These hamper the analysis and impose the
need to validate candidate causative variants by
Sanger sequencing. Moreover, WES analysis
imposes the need of applying filtering strategies.
This step is critical as it will limit data analysis and
consequently influence the results. For instance,
almost half of the variants may be excluded for
being synonymous. Despite the fact that these are
usually not considered deleterious, numerous
synonymous mutations have been implicated in
human diseases (Sauna & Kimchi-Sarfaty, 2011). In
addition, during WES analysis a frequency filter is
usually set to exclude variants with minor allele
frequency above a certain threshold (above 1% in
most cases). This threshold should be set in
accordance with the expected prevalence of the
disease. Even so, considering that in recessive
disorders carriers do not show any signs of the
disease, the frequency of damaging alleles in
populational variant databases can still be higher
than the established threshold. This would lead to
the erroneous exclusion of this variant during
filtering.
Homozygosity Mapping using Whole-Exome Sequencing: A Valuable Approach for Pathogenic Variant Identification in Genetic Diseases
211

Table 1: List of bioinformatic tools developed for ROH detection and their main features.
Software OS UI Algorithm
Main features /
experimental
design
Input
data
files
ROH
size
range
(Mb)
Other features Ref.
Homozygosity-
Mapper
Unix/Li
nux
(web
server
[a])
GUI
Detection of
homozygous
blocks of
selectable length
Perform
autozygosity
mapping from
SNP arrays and
NGS data
VCF
files
and
SNP
geno-
types
> 1.5
• Independent of
parameters like family
structure/allele
frequencies.
• Robust against
genotyping errors.
• Integrated with
GeneDistiller (candidate
gene search engine).
Seelow et
al., 2009
PLINK
Unix/Li
nux,
Mac
OS,
Win.
CLI &
GUI
Sliding-window
WGAS
analysis tool
set. Estimation
and use of IBD
in the context
of population-
based studies
BED,
PED,
and
FAM
files
[0.5 -
1.5]
> 1.5
• Makes a variety of
standard association tests.
• Maps disease loci that
contain multiple rare
variants in a population-
based linkage analysis.
• Integrated with
Haploview.
Purcell et
al., 2007
GERMLINE
Unix/Li
nux
CLI Sliding-window
Designed for
genome-wide
discovery of
IBD segments
shared within
large
populations
(SNP arrays)
PED,
MAP
and
Hap-
map
files
> 1.5
• Overcomes the
computational barrier of
pairwise analysis and can
scale the analysis linearly
with the sample size.
Gusev et
al., 2008
HomSI
Unix/Li
nux,
Mac
OS,
Win.
GUI Sliding-window
Identify ROH
in consangui-
neous families
from NGS data
VCF
files
> 1.5
• Takes into account
the distribution of the
variants within genomic
coordinates.
• Reported to be
consistent with data
derived from SNP
microarrays.
Gormez
et al.,
2014
H
3
M
2
Unix/Li
nux
CLI
Heterogeneous
hidden Markov
model
Analyse
medium and
short ROH
obtained from
WES data
BAM
files
< 0.5
[0.5-1.5]
> 1.5
• Reported to be more
accurate than GERMLINE
and PLINK, especially in
the detection of short and
medium ROHs.
Magi et
al., 2014
Agile-Genotyper
and
Agile-
VariantMapper
Win. GUI
User-controllable
visualization of
homozygous
regions
ROH analysis
WES data and
SNP
genotyping file
data
SAM;
tab-
delimite
d text
files
> 1.5
• AgileVariantMapper
uses the genotypes of all
positions found to be
polymorphic.
• AgileGenotyper
deduces genotypes from
positions previously found
to be polymorphic in the
1000 Genomes Project
data set.
Carr et
al., 2013
Footnote: [a]-http://www.homozygositymapper.org; CLI- command-line interface; GUI- graphical user interface; NGS-
Next-generation sequencing; OS- operative system; Ref.- references; ROH- runs of homozygosity; SNP- single nucleotide
polymorphism; UI- user interface; VCF- variant call format; WES- whole-exome sequencing; WGAS- whole-genome
association studies; Win.- Windows.
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
212

3.1 Homozygosity Mapping using WES
Data
Recent studies have clearly demonstrated the power
and the effectiveness of applying homozygosity
mapping to WES data, in an attempt to identify
causative genes for Mendelian disorders (Gillespie
et al., 2014; Shamseldin et al., 2015). This approach
has the advantage of unraveling the causal variant
irrespective of the gene involved. The homozygosity
map allows narrowing down of the target data sets;
examination at the base-pair level then enables
identification of candidate causative variants.
This approach would start by mapping WES
reads against a reference genome (human hg19 in
our examples). Data derived from whole-genome
and RNA sequencing can also be used. This step is
usually performed through a bioinformatic pipeline
that generates SAM/ BAM (Sequence /Binary
Alignment Map) and, at the end of the process, a
variant call format (VCF) file. These files are then
used as input for homozygosity mapping analysis.
The position and zygosity of the obtained sequence
variants can be used to retrieve/infer ROH regions.
As all bioinformatic approaches, there is an error
rate associated that is difficult to estimate, since it is
highly dependent on the WES metrics and the
sequencing platform used.
Table 1 lists some of the tools available to
perform ROH. For instance, the web-based tool
HomozygosityMapper (Seelow & Schuelke 2012),
allows users to interactively analyze NGS data for
homozygosity mapping. Furthermore, PLINK
(Purcell et al., 2007) and GERMLINE (Gusev et al.,
2008), originally developed for the analysis of SNP
array data, are tools based on sliding-window
algorithms. In a sliding window analysis, the
statistics are calculated for a small frame of the data.
The window incrementally advances across the
region of interest and, at each new position, the
reported statistics are calculated. In this way,
chromosomes are scanned by moving a window of a
fixed size along their entire length and variation in
genetic markers across the region of interest can be
measured. This type of analysis reveals how
variation patterns change across a surveyed genomic
segment (Srinuandee & Satirapod 2015). EXome-
HOMozygosity is an example in which a sliding-
window algorithm (PLINK) is applied for WES-
based ROH detection (Pippucci et al., 2011).
However, the sliding-windows approaches
cannot be used easily with short/medium ROH sizes.
In order to solve this issue, Magi et al proposed a
new algorithm, H
3
M
2
, that is capable of detecting
smaller ROH (Magi et al., 2014). AgileGenotyper
(Carr et al., 2013) and HomSI (Gormez et al., 2014)
are other tools that can be used for the graphical
visualization of ROHs.
3.2 Case Studies
The following examples are shown to elucidate the
relevance and limitations of WES-based ROH. Two
patients listed in Table 1 have recessive conditions
caused by homozygous pathogenic variants
identified by WES. There was no indication of
parental consanguinity, and patient P2 had a sibling
affected by the same condition.
In both cases, retrospective analysis was based
on genome-wide homozygosity mapping performed
using HomozygosityMapper algorithm.
Data obtained from patient P1 revealed the
presence of two long stretches of homozygous SNPs
in chromosomes 1 and 17 (with 19 and 17 Mb,
respectively) (Figure 1, top panel). The affected
gene (CCDC103) is located in one of these regions,
specifically in chromosome 17. In this example the
proposed approach would be suitable to generate a
considerably shorter list of candidate loci, and
consequently reduce the number of variants to be
evaluated in terms of their pathogenicity. In the
second example the same strategy was applied in a
patient with a rare neuromuscular disease.
Table 2: Cases selected to illustrate the use of homozygosity mapping using WES data in patients with rare genetic diseases.
Patient Phenotype Genotype Reference
P1, male adult
Infertility due to total sperm immotility.
Clinical features compatible with Kartagener
syndrome. No known parental consanguinity.
Homozygous pathogenic
variant in CCDC103 gene:
Chr17(h19):g.42978470G>C
Pereira et al., 2015.
P2, female
neonate
Severe neuromuscular disease, congenital
hypotonia, respiratory distress and bone
fractures. No known parental consanguinity.
Homozygous pathogenic variant in
ASCC1 gene:
Chr10(hg19):g.73970545dupC
Oliveira et al.,
(submitted).
Homozygosity Mapping using Whole-Exome Sequencing: A Valuable Approach for Pathogenic Variant Identification in Genetic Diseases
213
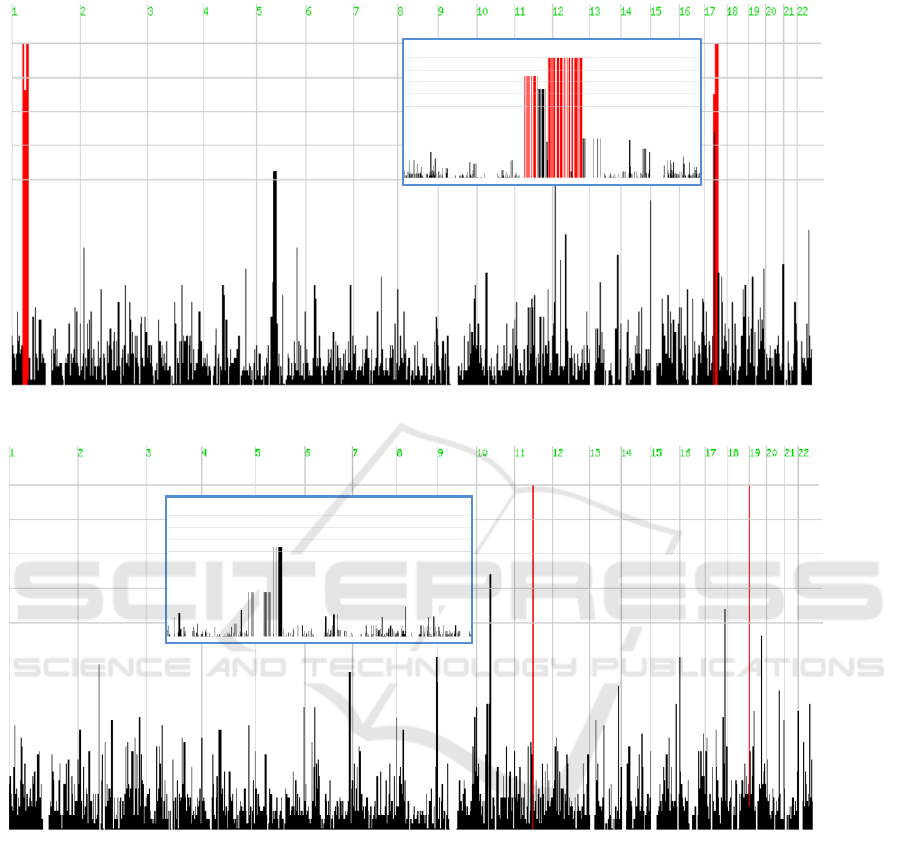
P1
P2
Chr. 10
Chr. 17
1.0 x max
0.9 x max
0.8 x max
0.7 x max
0.6 x ma
x
1.0 x max
0.9 x max
0.8 x max
0.7 x max
0.6 x max
Figure 1: Genome-wide homozygosity mapping using WES and the HomozygosityMapper software. Results are shown for
patients P1 and P2. Longer ROH are shown in red color. Inserted blue boxes show the genomic regions in more detail
where the disease-related gene is located.
Results for patient P2 revealed two smaller ROH
(1.8 and 0.2 Mb) in chromosomes 11 and 19
respectively (Figure 1, bottom panel). However, the
ASCC1 gene, carrying the disease-causing variant, is
located in chromosome 10. In this last example, and
considering the algorithm used, the analysis based
on the assumption of a homozygous variant located
in a ROH would clearly fail. The causal gene would
not be included in the list of candidate genes, most
likely due to the size of the actual ROH where the
genetic defect is located. In the first example, the
long ROH can be attributed to consanguinity
(although it was not formally confirmed), while in
the second case, a distant common ancestor could
explain the presence of a rare pathogenic variant in a
smaller ROH tract.
4 CONCLUSIONS
The aim of this position paper is to revisit
homozygosity mapping as an important tool for
clinical genetics in cases where a recessive disease is
suspected. We have reviewed the proposed
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
214

algorithms and available bioinformatic tools
designed for ROH detection based on WES data.
Selection of appropriate algorithms should mainly
consider specific features of the case under study,
such as the genetic context, the ROH size and the
number of relatives affected by the same condition.
Monogenic disorders have been studied by
classical approaches aiming to unravel several
disease-causing genes. The presence of numerous
genes in a candidate genomic region was a limiting
factor, considering the costs and the time required,
to screen mutations by Sanger sequencing. Another
limitation with the “traditional” approaches is more
evident when they are used to study trios (only the
parents and their affected child) or larger families
with only one or two affected members. The
genotype data extracted in these familial contexts
generally remain statistically insufficient for
classical analytical approaches (Jorde, 2000).
As compared with conventional homozygosity
mapping that uses known SNPs, WES has the added
advantage of allowing the identification of the actual
disease-causing variant. Therefore, instead of using
two different procedures, one for identifying
candidate loci and the other to identify the genetic
defect itself at the nucleotide level, both can be
performed in a single step by WES. Nonetheless,
there are still limitations and further bioinformatic
developments are required. Considering the
examples presented, there are sensitivity issues
especially if the genetic defect is in a small ROH.
Finally, we consider that it would be useful to
develop a bioinformatic tool that combines variant
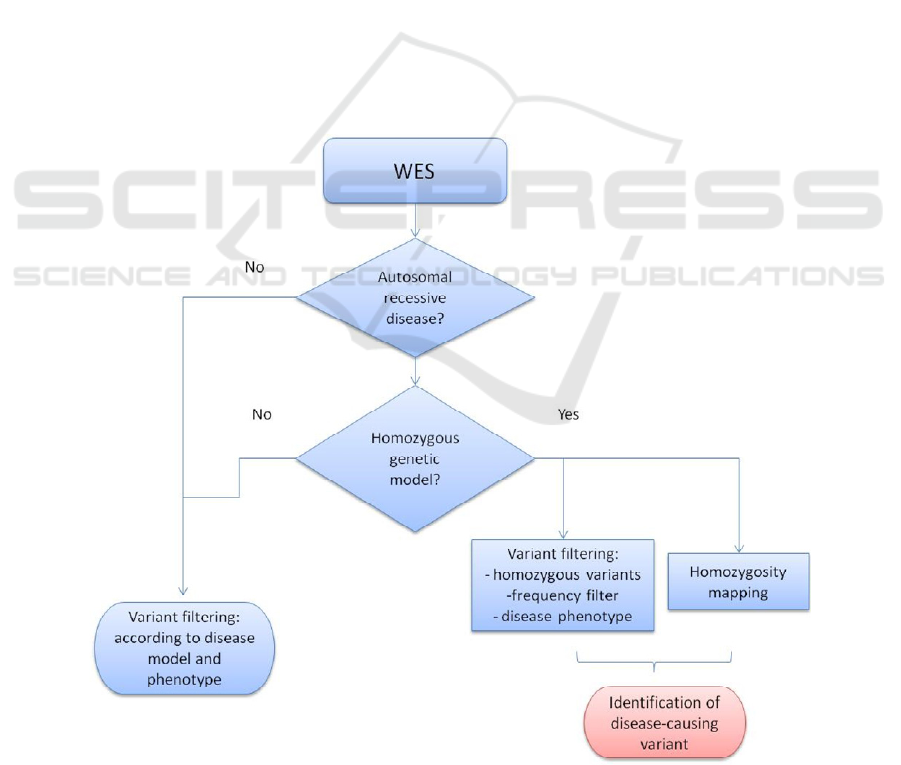
filtering and homozygosity mapping (Appendix),
which currently need to be performed separately.
ACKNOWLEDGEMENTS
The authors acknowledge support from: i) Fundação
para a Ciência e Tecnologia (FCT) [Grant ref.:
PD/BD/105767/2014] (R.P.); ii) Research grant
attributed by “Fundo para a Investigação e
Desenvolvimento do Centro Hospitalar do Porto”
[Grant ref.: 336-13(196-DEFI/285-CES)] (J.O.). The
work was also supported by the Institutions of the
authors and in part by UMIB, which is funded by
through FCT under the Pest-OE/SAU/UI0215/ 2014.
The authors would like to thank the clinicians for
patient referral.
REFERENCES
Abecasis, G. R. et al., 2002. Merlin—rapid analysis of
dense genetic maps using sparse gene flow trees.
Nature Genetics, 30(1), pp.97–101.
Alkuraya, F. S., 2010. Homozygosity mapping: One more
tool in the clinical geneticist’s toolbox. Genetics in
Medicine, 12(4), pp.236–239.
Bittles, A. H., 2001. Consanguinity and its relevance to
clinical genetics. Clinical genetics, 60(2), pp.89–98.
Boycott, K. M. et al., 2013. Rare-disease genetics in the
era of next-generation sequencing: discovery to
translation. Nat Rev Genet, 14(10), pp.681–691.
Carr, I. M. et al., 2013. Autozygosity Mapping with
Exome Sequence Data. Human Mutation, 34(1),
pp.50–56.
Christianson, A., Howson, C.P. & Modell, B., 2006.
Global report on birth defects: the hidden toll of dying
and disabled children, New York. Available at:
http://www.marchofdimes.org/materials/global-report-
on-birth-defects-the-hidden-toll-of-dying-and-
disabled-children-full-report.pdf [Accessed November
15, 2016].
Gillespie, R. L., Lloyd, I. C. & Black, G. C. M., 2014. The
Use of Autozygosity Mapping and Next-Generation
Sequencing in Understanding Anterior Segment
Defects Caused by an Abnormal Development of the
Lens. Human Heredity, 77(1–4), pp.118–137.
Goodship, J. et al., 2000. Report Autozygosity Mapping of
a Seckel Syndrome Locus to Chromosome 3q22.1-
q24. Am. J. Hum. Genet, 67, pp.498–503.
Gormez, Z., Bakir-Gungor, B. & Sagiroglu, M. S., 2014.
HomSI: a homozygous stretch identifier from next-
generation sequencing data. Bioinformatics, 30(3),
pp.445–447.
Gudbjartsson, D. F. et al., 2000. Allegro, a new computer
program for multipoint linkage analysis. Nature
Genetics, 25(1), pp.12–13.
Gusev, A. et al., 2008. Whole population, genome-wide
mapping of hidden relatedness. Genome Research,
19(2), pp.318–326.
Jorde, L. B., 2000. Linkage disequilibrium and the search
for complex disease genes. Genome research, 10(10),
pp.1435–1444.
Kruglyak, L. et al., 1996. Parametric and Nonparametric
Linkage Analysis: A Unified Multipoint Approach.
Am. J. Hum. Genet, 58, pp.1347–1363.
Magi, A. et al., 2014. H3M2: detection of runs of homozy-
gosity from whole-exome sequencing data. Bioinfor-
matics (Oxford, England), 30(20), pp.2852–2859.
McQuillan, R. et al., 2008. Runs of homozygosity in
European populations. American journal of human
genetics, 83(3), pp.359–372.
Ng, S. B. et al., 2010. Exome sequencing identifies the
cause of a mendelian disorder. Nature Genetics, 42(1),
pp.30–35.
Oliveira, J. et al., 2015. New splicing mutation in the
choline kinase beta (CHKB) gene causing a muscular
dystrophy detected by whole-exome sequencing.
Journal of Human Genetics. pp. 305-312.
Homozygosity Mapping using Whole-Exome Sequencing: A Valuable Approach for Pathogenic Variant Identification in Genetic Diseases
215
Oliveira, J. et al. 2017. Confirmation of a neuromuscular
phenotype related with defects in the ASC-1 complex:
report of the second case due to ASCC1 pathogenic
variants. Submitted.
Pemberton, T.J. et al., 2012. Genomic patterns of
homozygosity in worldwide human populations. The
American Journal of Human Genetics, 91(2), pp.275–
292.
Pereira, R. et al., 2015. Mutation analysis in patients with
total sperm immotility. Journal of assisted
reproduction and genetics, pp.1–10.
Pippucci, T. et al., 2011. EX-HOM (EXome
HOMozygosity): A Proof of Principle. Human
Heredity, 72(1), pp.45–53.
Purcell, S. et al., 2007. PLINK: a tool set for whole-
genome association and population-based linkage
analyses. American journal of human genetics, 81(3),
pp.559–575.
Sauna, Z. E. & Kimchi-Sarfaty, C., 2011. Understanding
the contribution of synonymous mutations to human
disease. Nature Reviews Genetics, 12(10), pp.683–
691.
Seelow, D. et al., 2009. HomozygosityMapper--an
interactive approach to homozygosity mapping.
Nucleic acids research, 37(Web Server issue),
pp.W593-9.
Seelow, D. & Schuelke, M., 2012. Homozygosity
Mapper2012—bridging the gap between homozygo-
sity mapping and deep sequencing. Nucleic acids
research, 40(W1), pp.W516–W520.
Shamseldin, H.E. et al., 2015. Identification of embryonic
lethal genes in humans by autozygosity mapping and
exome sequencing in consanguineous families.
Genome Biology, 16(1), p.116.
Sirmaci, A. et al., 2012. Challenges in Whole Exome
Sequencing: An Example from Hereditary Deafness I.
Schrijver, ed. PLoS ONE, 7(2), p.e32000.
Sobel, E., Papp, J.C. & Lange, K., 2002. Detection and
Integration of Genotyping Errors in Statistical
Genetics. Am. J. Hum. Genet, 70, pp.496–508.
Srinuandee, P. & Satirapod, C., 2015. Use of genetic
algorithm and sliding windows for optimising
ambiguity fixing rate in GPS kinematic positioning
mode. Survey Review, 47(340), pp.1–6.
APPENDIX
Appendix: Integration of homozygosity mapping in the analysis workflow of whole-exome sequencing (WES).
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
216
