
On the Impact of Medical Device Regulations on Software Architecture
Klaus Marius Hansen and Konstantinos Manikas
Department of Computer Science, University of Copenhagen, Njalsgade 128, Copenhagen, Denmark
Keywords:
Medical Device, Regulation Compliance, Software Architecture.
Abstract:
Compliance to regulations and regulatory approval are requirements for many medical device software sys-
tems. In this paper, we investigate the implications of medical device software regulations to the design of
software systems. We do so by focusing on the American and European regulatory authorities and review
the legal requirements for regulatory approval of medical devices. We define a simplified process for regula-
tory approval, consisting of five steps, and enhance this process by descriptions of how to decide whether a
software system is a medical device and how to identify the class of the device. Moreover, we review soft-
ware modularity in the implementation of software medical device and propose a set of preliminary principles
for architectural design of software medical device based on a set of constrains identified from the reviewed
regulations.
1 INTRODUCTION
Information and Communication Technology (ICT)
and, more specifically, software systems have ar-
guably become an integral part of healthcare ser-
vices and support a wide variety of domains and
functions within healthcare. This has become more
apparent with trends like unified and interoperable
Electronic Medical Records (EMRs) (Aanestad and
Jensen, 2011), the increased use of telemedicine,
e-health and m-health solutions (Christensen et al.,
2014), or mobile apps with functionality that pose
risks to human safety (Manikas and Hansen, 2013).
Many healthcare systems are mission-critical sys-
tems for which a failure may have severe conse-
quences for human lives. To moderate issues of this
kind, several authorities have issued regulatory re-
quirements similar to those for traditional medical
equipment and pharmaceutical substances. In this pa-
per, we argue that the design of software systems un-
der regulation compliance differs from other systems
and identify the impact of regulation compliance on
software design and software architecture. To do so,
we focus on two regulatory authorities: the United
States (US) Food and Drug Administration (FDA) and
the European Union’s (EU’s) Medical Device Direc-
tive (MDD) and review their regulatory requirements.
Our findings include: (i) a summary of the regula-
tory process in five simple steps, (ii) specifying how
to identify if a system is a medical device in FDA and
MDD, (iii) steps for defining the class of the medi-
cal device in each authority, (iv) identifying the con-
straints regulations put on system modularity, and (v)
proposing three architectural principles for medical
device software.
This work serves as a review of the legal require-
ments for medical device regulation in US and EU.
We translate the formal requirements to a set of steps
to be taken as part of regulatory compliance. More-
over, we identify a set of constraints on architectural
design of software medical device and propose three
principles to be included in the design of these sys-
tems.
2 REGULATION PROCESS
Examining the regulatory approval process for medi-
cal devices, we simplify the process for software sys-
tems into five steps (Intertek, 2015):
1. Determine if the software system is a medical de-
vice
2. Classify the software system as a medical device
3. Prepare technical documentation and develop and
implement a Quality Management System (QMS)
4. Fulfill premarket requirements and apply for as-
sessment
5. Maintain QMS and perform post-market surveil-
lance
Hansen, K. and Manikas, K.
On the Impact of Medical Device Regulations on Software Architecture.
DOI: 10.5220/0005776803890394
In Proceedings of the 9th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2016) - Volume 5: HEALTHINF, pages 389-394
ISBN: 978-989-758-170-0
Copyright
c
2016 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
389
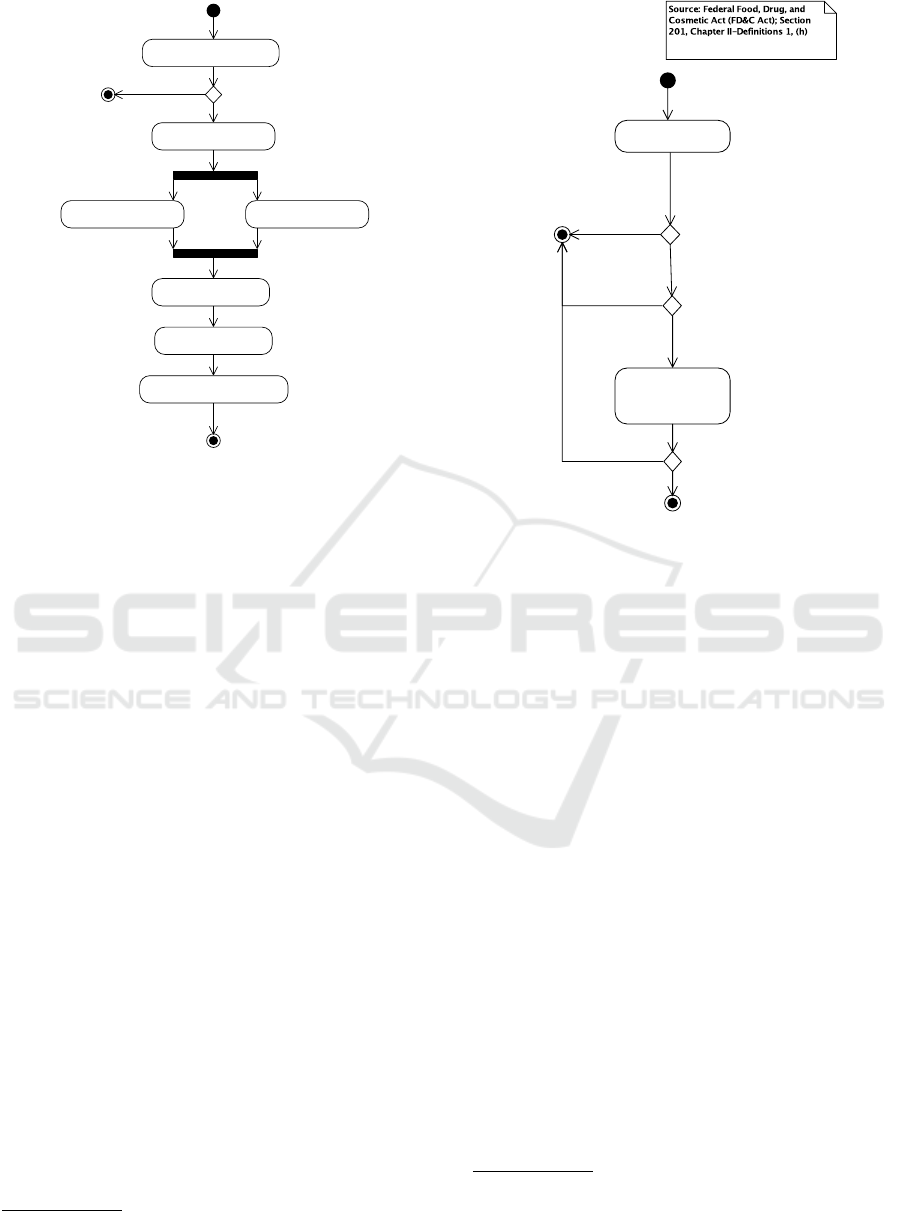
1. Determine if software meets the
definition of a "medical device"
2. Classify software as a
medical device
3a. Prepare technical
documentation
3b. Develop and implement
quality management system
4a. Fulfill premarket
requirements
4b. Apply for assessment
5. Maintain quality management
system and post-market surveillance
[yes]
[no]
Figure 1: Overview of the regulatory approval process for
medical devices (EU, US). Source: Intertek (2015).
Figure 1 shows the overview of the regulatory ap-
proval process
1
. Our main sources are the laws and
regulations that guide the approval and marketing of
medical devices. In the US, the Food and Drug Ad-
ministration (FDA) approves medical devices. The
FDA is governed by the Federal Food, Drug, and Cos-
metic Act (FD&C Act; (Federal Food, Drug, and Cos-
metic Act of 1938, 2007, Chapter 9)) that is codified
in the Code of Federal Regulations (CFR), Title 21
(FDA, 2014). In the EU, medical devices need to
meet the requirements of the Medical Devices Direc-
tive (MDD; MDD (2007)) to be put on market. Below
we examine steps 1 and 2 in more detail for FDA and
MDD.
2.1 Determine if Software is a Medical
Device
Step 1 in Figure 1 focuses on whether the software
under consideration is a medical device or not. If
the software is not a medical device, neither FDA nor
MDD regulations apply.
Figure 2 gives an overview for the US (based on
Section 201(h) of the FD&C act). Whether software
is a medical device is based on the “intended use” of
the software. Similarly, Figure 3 gives an overview
for the EU (based on the MDD; (MDD, 2007, Arti-
cle 1)). An important distinction is that in the US,
an accessory is also a medical device whereas in the
1
Although these steps can also be applied to Canada and
Japan, we focus on the regulatory processes of EU and US.
Determine intended use
of software
Determine if software is
component, part, or
accessory to a medical
device
Medical device
May not be a medical device
[no]
[yes]
[yes]
[intended to
affect the structure or
any function of the body]
[yes]
[intended for
diagnosis of disease or other
conditions, OR
in the cure, mitigation, treatment, or
prevention of disease]
Figure 2: Determining if software is a medical device, US.
EU, accessories are governed by the MDD but are not
themselves medical devices.
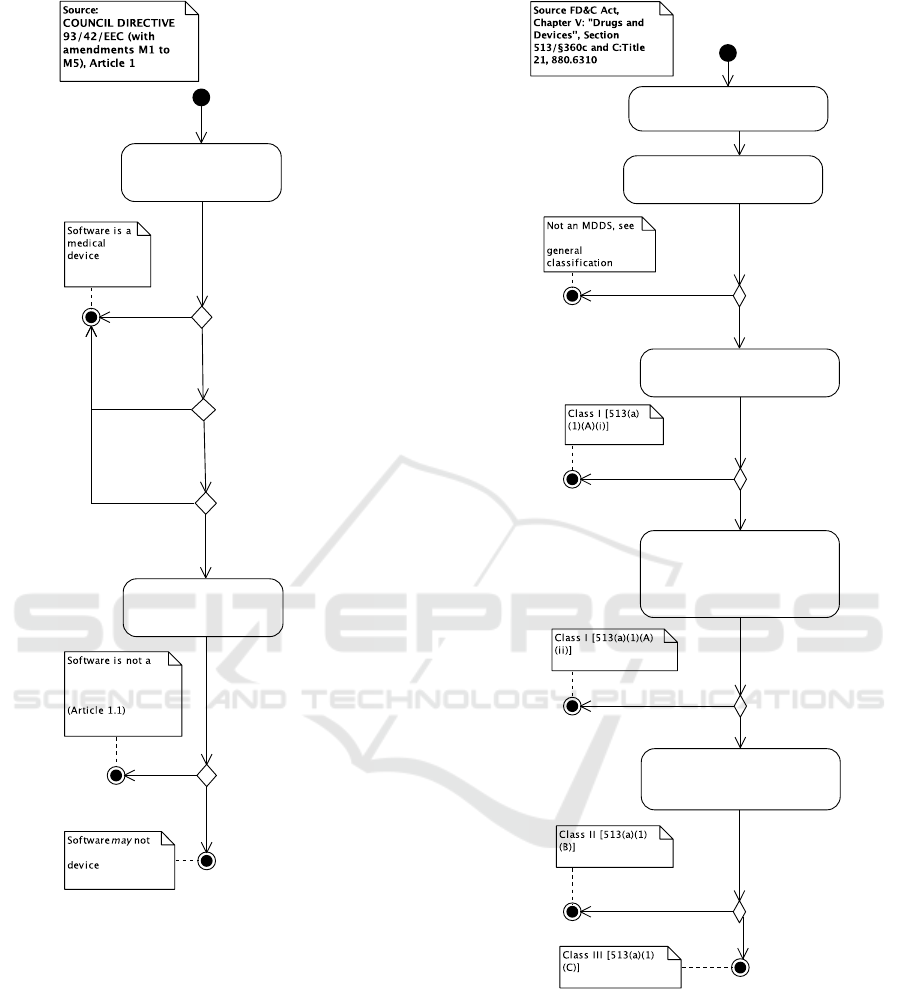
2.2 Classification of Software as
Medical Devices
Assuming that the software is a medical device, the
next step is to determine the classification category
the software falls under. FDA distinguishes between
three medical device classes: Class I, II, or III, while
MDD distinguishes four: Class I, IIa, IIb, or III. These
classifications imply requirements for the software
development process (including steps 3, 4, and 5 in
Figure 1).
The classification process of the US is shown in
Figure 4. It is important to note that the classification
found is only a suggestion and that it is the FDA that
determines the class of the software. In the US pro-
cess, the first step for classification is to find a device
that is “substantially equivalent” to the software. If
such a device can be found in the CFR, and an equiv-
alence argument can be made, the software gets the
same classification as the substantially equivalent de-
vice. Substantially equivalent devices may be sought
through FDA’s product classification database
2
. If no
2
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfP
CD/PCDSimpleSearch.cfm. The database currently
(2015-05-27) contains 103 matches for the search string
“software”.
HEALTHINF 2016 - 9th International Conference on Health Informatics
390
Determine intended use of
software
Determine if software is
accessory to a medical device
be a medical
medical device,
but falls under
MDD anyway
[no]
[Software is intended
specifically by its
manufacturer to be used
together
with a device to enable it to
be used in accordance with
the use of
the device intended by the
manufacturer of the device]
[yes]
[Intended use is investigation,
replacement or modification
of the anatomy or a physiological
process]
[yes]
[Intended use is diagnosis,
monitoring, treatment,
alleviation
of or compensation for an
injury or handicap]
[Intended use is
diagnosis, prevention,
monitoring, treatment or
alleviation of disease]
[yes]
Figure 3: Determining if software is a medical device, EU.
match is found, the rest of Figure 4 applies.
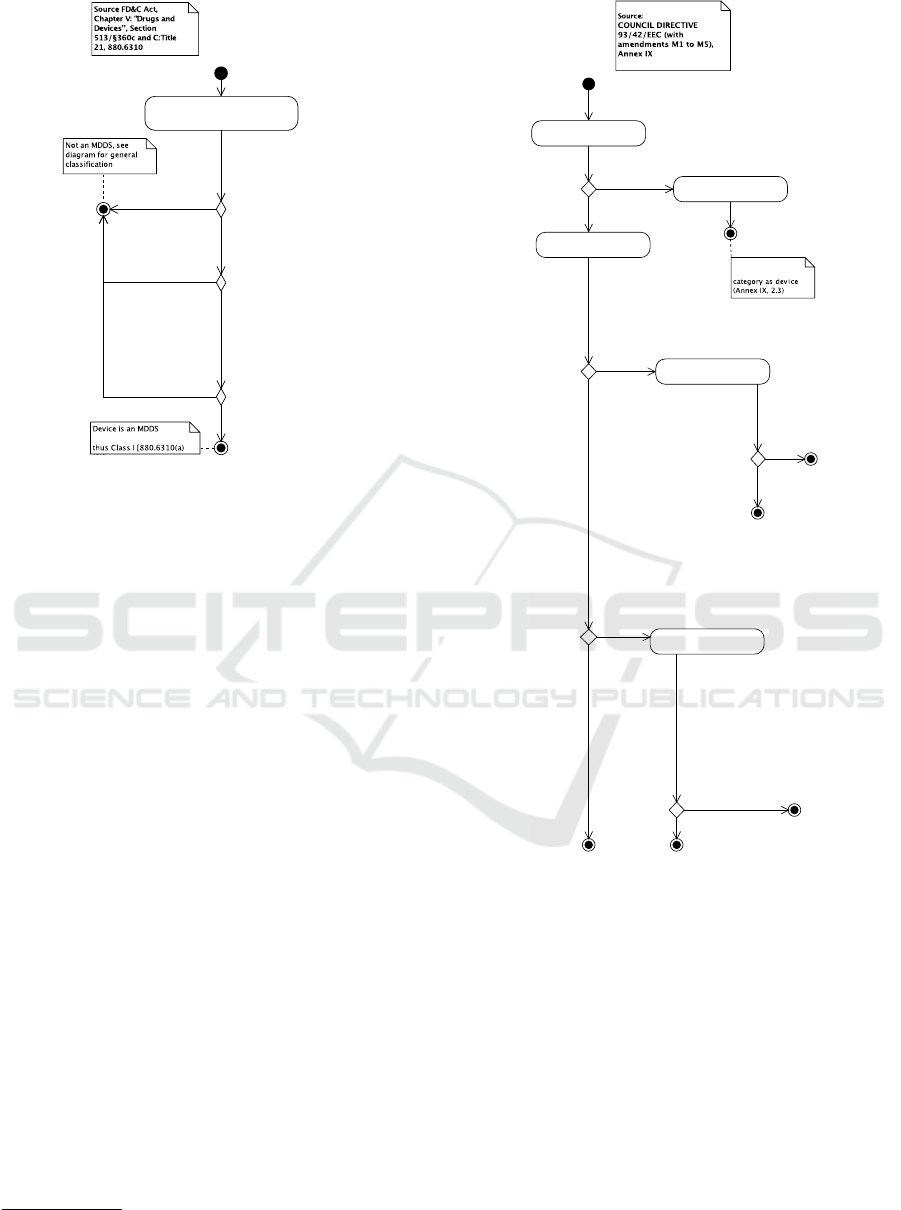
A special case of interest for software in the FDA
classification are the “Medical Device Data Systems
(MDDS)”. MDDS transfer, store, convert, or display
medical device data without controlling or altering the
functions of connected medical devices or being used
for active patient monitoring. MDDS fall under Class
I. This is shown in Figure 5. Food and Drug Admin-
istration (2011) contains detailed questions and an-
swers related to MDDS.
The EU classification process is shown in Fig-
ure 6 (MDD, 2007, Annex IX). In the EU, software
that “drives or influences the use of” a device is clas-
diagram for
Identify existing device that is
"substantially equivalent" [513(i)(1)] to
software
Determine if general controls are
sufficient for reasonable assurance of
safety and effectiveness
Insufficient information to determine,
but not supporting or sustaining
human life or preventing impairment of
human health and does not represent a
potential unreasonable risk of illness or
injury
Determine if special controls are
sufficient to provide reasonable
insurance of safety and effectiveness
Determine intended use, conditions of
use, benefit/risk of software [513(a)(2)]
[yes]
[no]
[yes]
[yes]
[no]
[no device found]
[yes]
Figure 4: Classifying medical device software, US.
sified as the device is. Otherwise, software is consid-
ered a “standalone, active device” and can be classi-
fied in Class I, IIa, or IIb. In particular, according to
Figure 6, software that is a standalone, active device,
and an MDDS is in Class I.
On the Impact of Medical Device Regulations on Software Architecture
391
Determine intended use, conditions of
use, benefit/risk of software [513(a)(2)]
[880.6310(a)(1)] and
´
[yes]
[no]
[Intended for
(i) The electronic transfer of
medical device data;
(ii) The electronic storage of
medical device data;
(iii) The electronic conversion of
medical device data from one
format to another format in
accordance with a preset
specification; or
(iv) The electronic display of
medical device data]
[yes]
[intended to be used in
connection with active
patient monitoring
[880.6310(a)(2)]]
[intent is controlling or
altering the functions or
parameters of any
connected medical devices
[880.6310(a)(1)]]
[yes]
Figure 5: Classifying medical device software, US. Special
case: Medical Device Data System (MDDS).
3 MODULARITY OF SOFTWARE
AS MEDICAL DEVICES
In this section we examine what constitutes “soft-
ware”, i.e., which parts of a (software) system is a
medical device. As noted in Sections 2.1 and 2.2 this
is important since restrictions apply for how software
medical devices may be implemented. The high-level
division of a software system is its “software architec-
ture”, i.e.,
“the set of structures needed to reason about
the system, which comprise software ele-
ments, relations among them, and properties
of both” (Bass et al., 2013)
As such, the module, component-and-connector
(C&C), and allocation structures are all relevant to
how a software medical device should and could be
divided into parts.
For the EU, guidance documents state that “the de-
termination of the class of a particular device may be
made with respect to the simplest configuration that
can still be considered, in view of its proper func-
tional features, as a device in its own right” (MED-
DEV 2.4/1 Rev. 9, 2010). MEDDEV 2.1/6 (2012)
discusses modularization of software medical devices
into separate applications including that non-medical
modules of a software medical device system is not
to be treated as a medical device
3
. Rather, such
3
This guidance relates to that accessories are “treated as
medical devices in their own right”(MDD, 2007).
Determine intended use of
software
Determine device classification
Software is standalone, active
device (Annex IX, 1.4)
Software classified
in the same
Determine whether software is
hazardous
Determine whether software is
hazardous
Class IIb (Rule 9)
Class IIa (Rule 9)
Class I (Rule 12)
Class IIb (Rule 10)
Class IIa (Rule 10)
[no]
[yes]
[monitoring of vital physiological
parameters, where the
nature of variations is such that it could
result in immediate
danger to the patient (Annex IX, Rule 10)
OR
intended to emit (or control or monitor
devices that do)
ionizing radiation and intended for
diagnostic and therapeutic
interventional radiology (Annex IX, Rule
10)]
[no]
[yes]
[yes]
[administer or
exchange energy to
or from
the human body in
a potentially
hazardous way
(Annex IX, Rule 9)]
[yes]
[Software is active device for diagnosis
(Annex IX, 1.6)
AND
(intended to supply energy which will
be absorbed by
the human body (Annex IX, Rule 10)
OR
intended to image in vivo distribution
of radiopharmaceuticals
(Annex IX, Rule 10)
OR
intended to allow direct diagnosis or
monitoring of vital
physiological processes (Annex IX,
Rule 10))]
[Software is active therapeutic
device (Annex IX, 1.5)
AND
intended to administer or exchange
energy
(Annex IX, Rule 9)]
[no]
[yes]
[drives a device or influences
the use of a device]
Figure 6: Classifying medical device software, EU.
non-medical modules are to be treated as “Software
Of Unknown Provenance” (SOUP; IEC 62304:2006
(2006)). Still “the whole combination [...] must be
safe and must not impair the specified performances
of the devices” (MDD, 2007).
For the US, guidance on modularity is less clear as
is the status of “accessories” (Food and Drug Admin-
istration, 2015) including their definition. For some
modularizations, a module may become an accessory
to a parent device/module. The guidance clarifies how
accessories are defined and how they may be clas-
sified (and suggests the use of the “De Novo” pro-
cess for classifying accessories in a lower class than
their parent device; Food and Drug Administration
(2015)).
HEALTHINF 2016 - 9th International Conference on Health Informatics
392

The IEC 62304:2006 (2006) standard is har-
monized and adopted by both the EU and the
US (Bundtz, 2010) and thus applies to both cases. The
standard specifies that a software medical device sys-
tem should first be classified as a whole based on a
hazard analysis (Bundtz, 2010). Subsequently, mod-
ules (“software items”) may be classified separately
if they are “segregated”. The standard only mentions
one example of segregation: “to have software items
execute on different processors. The effectiveness of
the segregation can be ensured by having no shared
resources between the processors”.
4 ARCHITECTURAL
PRINCIPLES FOR SOFTWARE
AS MEDICAL DEVICES
To decide on a proper modularization for software
medical devices, multiple constraints have to be taken
into account:
• The cost of putting a device to market increases
with increased classification. For PMAs, the
FDA, e.g., charges more than $250,000
4
. More-
over, for Class II or above devices in the EU, an
external, notified body has to assess conformity to
regulations.
• Time-to-market increases with increased classifi-
cation. The higher the classification, the higher
the requirements to plans, requirement specifi-
cation, software architecture design etc. (IEC
62304:2006, 2006).
• Evolvability decreases with increased classifica-
tion. For the US, e.g., changes to a Class III de-
vice may require a “PMA supplement” that may
take up to 180 days to process
5
.
On the other hand, if a device is marketed according
to a lower class than it really is, the consequences may
be that a device is taken off the market
6
.
We thus propose the following preliminary princi-
ples for architectural design:
• Form Equivalence Classes of Components
Through Segregation. Divide/segregate software
medical devices into modules that are separate
applications/devices or accessories with separate
classes if possible.
4
http://www.fda.gov/MedicalDevices/DeviceRegulation
andGuidance/Overview/MDUFAIII/ucm313673.htm
5
http://www.fda.gov/RegulatoryInformation/Guidances/
ucm089274.htm
6
http://www.fda.gov/ICECI/EnforcementActions/Warn
ingLetters/2013/ucm376296.htm
• Segregate Transforming Components from Trans-
mitting Component. (For the US) MDDS devices
are of particular interest since they are Class I.
• Segregate Evolving Components from Stable.
Components that are expected to evolve fre-
quently should present as little risk as possible,
i.e., be in a low class. In this way, e.g., adaptive
maintenance can be performed faster.
5 CONCLUSION
In this paper, we investigate the implications of med-
ical device software regulations to the design of soft-
ware systems. We do so by focusing on the US and
EU authorities and review the legal requirements for
regulatory approval of medical devices. We define a
simplified process for regulatory approval, consisting
of five steps, and enhance this process by steps that
aid in deciding whether a software system is a med-
ical device and how to identify the class of the de-
vice. Moreover, we review software modularity in
the implementation of software medical devices and
propose a set of preliminary principles for their archi-
tectural design based on a set of constraints identified
from the reviewed literature.
Plans for future work include the improvement
and empirical evaluation of the identified process and
steps in different domains of software systems. More-
over, we plan to apply and evaluate the proposed prin-
ciples for architectural design in practice.
ACKNOWLEDGEMENTS
This work has been conducted as part of the SCAUT
Project
7
, co-funded by the Innovation Fund Denmark.
REFERENCES
Aanestad, M. and Jensen, T. B. (2011). Building nation-
wide information infrastructures in healthcare through
modular implementation strategies. The Journal of
Strategic Information Systems, 20(2):161 – 176.
Bass, L., Clements, P., and Kazman, R. (2013). Software
Architecture in Practice. Addison-Wesley, 3rd edition.
Bundtz, B. (2010). Developing medical device software
to iec 62304. European Medical Device Technol-
ogy, 1(6). http://www.emdt.co.uk/article/developing-
medical-device-software-iso-62304.
7
http://www.scaut.dk/
On the Impact of Medical Device Regulations on Software Architecture
393

Christensen, H. B., Hansen, K. M., Kyng, M., and
Manikas, K. (2014). Analysis and design of software
ecosystem architectures – towards the 4s telemedicine
ecosystem. Information and Software Technology,
56(11):1476 – 1492.
FDA (2014). Title 21 of the code of federal regulations.
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cf
cfr/cfrsearch.cfm.
Federal Food, Drug, and Cosmetic Act of 1938 (2007).
United States Code, Title 21.
Food and Drug Administration (2011). Med-
ical devices; medical device data sys-
tems. Federal Register, 76(31):8637–8649.
http://www.gpo.gov/fdsys/pkg/FR-2011-02-
15/pdf/2011-3321.pdf.
Food and Drug Administration (2015). Medical device
accessories: Defining accessories and classification
pathway for new accessory types. draft guidance
for industry and food and drug administration staff.
http://www.fda.gov/downloads/medicaldevices/device
regulationandguidance/guidancedocuments/ucm4296
72.pdf.
IEC 62304:2006 (2006). Medical device software – soft-
ware life cycle processes. ISO/IEC.
Intertek (2015). Quick guide: 5 steps to market approval
for medical devices. http://www.intertek.com/5-step-
market-approval-medical-devices/. Accessed May
2015.
Manikas, K. and Hansen, K. M. (2013). Reviewing the
health of software ecosystems - a conceptual frame-
work proposal. In Alves, C. F., Hanssen, G. K., Bosch,
J., and Jansen, S., editors, Proceedings of the 5th In-
ternational Workshop on Software Ecosystems, Pots-
dam, Germany, June 11, 2013, volume 987, pages 33–
44.
MDD (2007). Council directive 93/42/eec
of of 14 june 1993 concerning medi-
cal devices. http://eur-lex.europa.eu/legal-
content/EN/TXT/PDF/?uri=CELEX:01993L0042-
20071011&from=EN. Amended up until and
including Directive 2007/47/EC.
MEDDEV 2.1/6 (2012). Guidelines on the qualification and
classification of stand alone software used in health-
care within the regulatory framework of medical
devices. http://ec.europa.eu/growth/sectors/medical-
devices old/documents/guidelines/files/meddev/2 1
6 ol en.pdf.
MEDDEV 2.4/1 Rev. 9 (2010). Medical devices:
Guidance document – classification of medi-
cal devices. http://ec.europa.eu/health/medical-
devices/files/meddev/2 4 1 rev 9 classification en.pdf.
HEALTHINF 2016 - 9th International Conference on Health Informatics
394
