
Prediction of Cancer using Network Topological Features
Fernanda Brito Correia
1,3
, Joel P. Arrais
2
and José Luís Oliveira
1
1
Dept. of Electronics, Telecommunications and Informatics (DETI), Inst. of Electronics and Informatics Engineering of
Aveiro (IEETA), University of Aveiro, Aveiro, Portugal
2
Dept. of Informatics Engineering (DEI), Centre for Informatics and Systems of the University of Coimbra (CISUC),
University of Coimbra, Coimbra, Portugal
3
Dept. of Informatics and Systems Engineering (DEIS), Polytechnic Institute of Coimbra, Coimbra, Portugal
Keywords: Protein-protein Interaction Networks, Classification, Cancer Prediction.
Abstract: Several data mining methods have been applied to explore biological data and understand the mechanisms
that regulate genetic and metabolic diseases. The underlying hypothesis is that the identification of signatures
can help the clinical identification of diseased tissues. Under this principle many different methodologies have
been tested mostly using unsupervised methods. A common trend consists in combining the information
obtained from gene expression and protein-protein interaction networks analyses or, more recently, building
series of complex networks to model system dynamics. Despite the positive results that these works present,
they typically fail to generalize out of sample datasets. In this paper we describe a supervised classification
approach, with a new methodology for extracting the network topology dynamics embedded in a disease
system, to improve the capacity of cancer prediction, using exclusively the topological properties of biological
networks as features. Four microarrays datasets were used, for testing and validation, three from breast cancer
experiments and one from a liver cancer experiment. The obtained results corroborate the potential of the
proposed methodology to predict a certain type of cancer and the necessity of applying different classification
models to different types of cancer.
1 INTRODUCTION
Cancer is a complex genetic disease that affects an
increasing number of citizens all over the world. In
2015, more than 1.6 million new cancer cases are
expected in the United States, from which around
15% correspond to breast cancer (Siegel et al., 2015).
Understanding the underlying biological mechanisms
behind this disease has been the goal of many and
continuous research initiatives.
One strategy to study cancer is using microarrays,
a high-throughput technology that measures gene
expression, allowing the parallel analysis of genes in
several samples. Different individuals or different
conditions (healthy and non-healthy cells of the same
individual) originate distinct microarray samples with
gene expression values. These different samples can
reveal signatures that help to distinguish cancer from
non-cancer tissues.
In a network-based approach, bio-entities such as
genes and proteins can be represented as nodes and
their relationships as edges. Using this approach we
can model biological processes that can be analysed
using graph and network methods. The construction
of network-based models to study complex
phenomena, like cancer diseases, allows the capture
of the embedded systems dynamics. This dynamics
can be captured by constructing series of different
complex networks through time, through different
stages and through different traits. The study of the
topological properties of these complex networks
allows understanding specific structures, signatures
and similarities.
This paper presents a methodology to construct
protein-protein interaction networks to capture the
existent system dynamics beneath their topology.
These networks, here named Sample-Series
Networks (SSN), are constructed using a group of
cancer and healthy microarray samples. Information
features are exclusively obtained through the analysis
of the topological properties of these networks and
without any other biological information. Using the
obtained set of topological features a supervised
approach is then used to classify between cancer and
non-cancer tissues samples. This work aims to
Correia, F., Arrais, J. and Oliveira, J.
Prediction of Cancer using Network Topological Features.
DOI: 10.5220/0005696202070215
In Proceedings of the 9th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2016) - Volume 3: BIOINFORMATICS, pages 207-215
ISBN: 978-989-758-170-0
Copyright
c
2016 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
207

address several questions, namely: are there
evidences of signatures beneath the SSN that allow us
to classify samples as cancer or non-cancer? Which
topological measures give better results as
classification features among the several groups
considered? Does a classification model distinguish
different types of cancers?
Tests have been made in four gene expression
microarray experiments, three from breast cancer and
one from liver cancer.
2 BACKGROUND
Anomalies in a gene, protein or other bio-entity can
cause diseases and since the arrival of the next-
generation sequencing (NGS), that are found more
evidences of human genes being correlated to
diseases. Data from November 6
th
, 2015 obtained in
the Online Mendelian Inheritance in Man database
(OMIM) (Amberger et al., 2015) shows that there are
5597 phenotypes for which the molecular basis is
known and 3453 genes with phenotype-causing
mutation. Most disease genes are not essential, being
essential genes typically organized as hubs in a
complex network (Barabási et al., 2011, Jonsson and
Bates, 2006).
Biological networks are not random, they have
clustered groups of bio-entities, like genes or
proteins. They are also sparsely connected, which is
considered an evolutionary advantage for preserving
robustness to random failures (Barabasi and Oltvai,
2004). Also, it is known that genes and proteins that
are involved in the same phenotype are network
neighbours (Oti et al., 2006) and that a disease
phenotype can be associated to interactions in a
biological complex network that models these
biological processes (Menche et al., 2015).The
comparison of networks can use global and local
topological measures. Both are used in (Pržulj et al.,
2004) to show that the structure of yeast PPI networks
is closer to the geometric random graph model
relatively to graphlet frequency. In (Pržulj, 2007) a
new network similarity measure is defined based on
the graphlet degree distribution as a generalization of
the degree distribution.
Genomic changes that are translated to proteins
can alter biological functions and a system-based
approach modelled through complex networks can
assist the discovery of signatures related to disease
mechanisms, by analysing their topology (Vidal et al.,
2011, Barabási et al., 2011, Arrais and Oliveira, 2011,
Farkas et al., 2011).
Cliques help to understand the mechanisms
involved in cancer, since they are fully connected
subnetworks more conserved in biological networks.
In cliques, genes are functionally related and highly
expressed. In (Pradhan et al., 2012) it is proposed a
topological and biological feature-based network
approach, integrating the expression data, along with
network topological information and biological
information. Cliques are scored based on these
information and are considered as gene signatures for
the colorectal cancer (CRC).
Sets of biological complex networks can be
constructed across multiple conditions, like species,
time, and evolutionary states, traits or even samples,
as the novel approach used in this paper, building
dynamic models of the studied system.
A systems biology approach can be used to
interpret biological data. The (Trapé and Gonzalez-
Angulo, 2012) review addresses the contributions of
systems biology. DNA, RNA and protein changes
data are integrated to understand breast cancer
metastasis process. (Sonachalam et al., 2012) shows
how to build a PPI network representative of the
colorectal cancer (CRC) where nodes are
genes/proteins obtained from Gene Set Enrichment
Analysis (GSEA). (Barter et al., 2014) compares
single-gene, gene-set and two PPI network-based
methods, using gene expression microarrays data,
applied to melanoma and ovarian cancer. In single-
gene, features are the expression values of
informative genes identified via differential
expression analysis. In the gene-set method, genes are
grouped into sets using biological knowledge, which
are used as features for classification. In the first
network-based method features are the most
informative individual genes selected using the PPI
network, while in the second network-based method,
features are identified or are extracted from them
considering the edges or are sub-networks hub genes.
Three classifiers were used, namely Random Forest
(RF), Diagonal Linear Discriminant Analysis and
Support Vector Machines (SVM), with 5-fold cross
validation. It concludes that including network
information may lead to the identification of more
stable gene expression signatures.
(Dominietto et al., 2015), shows how to integrate
imaging data into networks to define tumor
fingerprints, through both network topology and the
detection of dynamic connectivity patterns.
In (Chuang et al., 2007) PPI subnetwork markers
are found to distinguish between metastatic and non-
metastatic tumors, using a score function. Candidate
subnetworks are built starting with a single protein
and are expanded using the PPI network, until the
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
208

score stops to increase. The activity scores calculated
from the average of the expression levels of each
subnetwork were used as feature values. The
classifiers used were based on logistic regression and
SVM using 5-fold cross validation. In (Chen and
Yang, 2014), normal, benign and malignant states of
breast cancer are differentiated, building a gene
regulatory network representative of each state and
comparing their network topological properties (in
and out-degree, betweenness, cluster coefficient and
closeness). Gene ranking was made selecting 53 hub
genes. (Wang et al., 2015) review describes pathway
and network-based approaches applied to cancer
biomarker discovery, in particular to the liver and
hepatocellular carcinoma (HCC). In (Ou-Yang et al.,
2014) dynamic PPI networks are constructed from
time-course gene expression data and PPI data,
extracting stable and dynamic interactions along time
to predict temporal protein complexes. An approach.
using differential co-expression analysis and PPI
networks for study human HCC progression that uses
subnetworks for each of the five stages of this
carcinoma can be found in (Yu et al., 2013).
Data mining classification techniques have been
used to look for signatures in cancer diseases. A large
number of variables can be used to characterize
cancer and non-cancer biological datasets, so it is
necessary to choose the most relevant. There are
several feature selection algorithms and a review is
presented in (Saeys et al., 2007), including the
ReliefF feature selection method (Kononenko, 1994)
used in this paper. In (Nancy and alias Balamurugan,
2013), the ReliefF feature selection method is
claimed to be the best method among several tested
for cancer classification using gene expression data.
Also ReliefF algorithm is efficient, and adequate
when there is much feature interaction, ranking well
the quality of features when there is a strong
dependency between them (Robnik-Šikonja and
Kononenko, 2003).
In (Furey et al., 2000), a score is calculated for the
expression values of genes, to select those with
highest scores as features in the classification
withhold-one-out cross validation. Tests were made
and the best results were obtained with 50 genes.
(Ramani and Jacob, 2013) uses a Bayesian Network
Learning (BNL) prediction to classify lung cancer
tumors as Small Cell Lung Cancer (SCLC), Non-
Small Cell Lung Cancer (NSCLC) and COMMON
classes, using the structural and physicochemical
properties of protein sequences obtained from genes
using microarray analysis. Several feature selection
methods were used with different prediction
techniques. Best results were obtained using BNL
with Gain Ratio. A model for predicting the survival
rate of patients affected by lung cancer, applying
different feature selection algorithms, can be found in
(Dezfuly and Sajedi, 2015). The classification
algorithms used were: Decision Tree (DT), BNL and
Neural Network (NN).
A new network-based supervised classification
method to predict cancer, named NBC and using only
gene expression levels is presented in (Ay et al.,
2014). It was applied to different datasets, (lung,
breast, leukaemia and colon cancers) using five
classification algorithms, namely SVM, KNN, NBL,
C4.5 and RF with 10-fold cross validation and with
five feature selection methods. A gene-association
network was created for each class, where nodes are
genes and edges represent the correlation between
their expression levels. High accuracy classification
was obtained with less than 100 genes.
3 METHODS
This paper describes a system-based approach to
classify between cancer and non-cancer tissues and
can contribute to find signatures that distinguish
disease biological processes from healthy biological
processes, using the topological properties of
networks and considering the network topological
dynamics embedded in the disease system.
A network-based method is used, by constructing
a set of PPI networks, one for each sample belonging
to the SSN. The ReliefF algorithm (Kononenko,
1994) is used to rank a subset of genes. In each SSN
network, nodes are proteins coded by a subset of the
most expressed genes of the top ReliefF genes and
edges indicate that the proteins coded by those genes
interact physically. A score was used as a threshold
for the PPI interactions.
Network topological properties were used as
features in the supervised binary classification
methodology and their values were obtained from the
topological analysis of each SSN network.
These classification models were evaluated using
the statistical measures, accuracy, precision, recall,
F1-score and area under the ROC curve.
Four datasets were used, three from breast cancer
microarray experiments and one from a liver cancer
microarray experiment. Three types of tests were
made: using 5 fold cross-validation; using data
obtained from two of the breast cancer datasets as
train set, and data obtained from the other breast
cancer dataset as test set; and using data obtained
from the three breast cancer datasets to train the
Prediction of Cancer using Network Topological Features
209

dataset and, for testing, using data obtained from the
liver cancer dataset.
3.1 Gene Expression Microarray Data
Sets
The experiments were obtained from ArrayExpress
(http://www.ebi.ac.uk/arrayexpress/): E-GEOD-
65194 (178 samples, where 167 are from breast
cancer tissue cells), E-GEOD-54002 (433 samples,
where 417 are from breast cancer tissue cells), E-
GEOD-29044 (124 samples, where 75 are from breast
cancer tissue cells) and E-MTAB-950 (276 samples,
where 179 are from liver cancer tissue cells). To
assure probes and samples uniformity all experiments
share the same array design A-AFFY-44 and all
samples were labelled as belonging to one of the two
classes, Cancer or Healthy. The 54673 genes of the
experiments were sorted by decreasing values of
ReliefF (Kononenko, 1994), an algorithm that can be
applied to continuous and discrete values. For each
experiment the top ReliefF 100 genes were selected
and merged in one matrix of 735 samples and 276
genes for breast cancer and one matrix of 124 samples
and 276 genes for the liver cancer. The PPI SSN was
obtained from the 100 most expressed genes from
each sample of the previous matrixes. The number
100 genes belongs to the typical interval of 50 to 150
number of genes used for binary classifications
studies (Ay et al., 2014).
3.2 Protein-protein Interaction
Networks
DAVID and UNIPROT were primarily used (Dennis
et al., 2003, Consortium, 2014) to obtain the mapping
of identifiers from probeset ids and gene names to
proteins. The human PPI dataset was obtained from
STRING, an online database resource with several
distinct types and sources of PPI information. Using
this dataset, several networks were constructed, one
for each sample, representing the entire set of PPI for
all different samples. Only PPIs with score greater or
equal to 300 were considered. These networks were
constructed as undirected, unweight and with no self-
edges.
3.3 Classification Methods
The set of supervised learning algorithms used were
the KNN classifier, the SVM classifier implemented
using an RBF kernel, and the RF, all with default
parameters.
Classification results were obtained using Orange
through scripting in Python and through visual
programming in Orange Canvas (Demsar et al.,
2007).
The statistical measures used to evaluate the
performance of the binary classification models were,
the classification accuracy (CA), the precision
(Precision), the recall (Recall), the F1-score (F1) and
the area under the ROC Curve (AUC) (Sokolova and
Lapalme, 2009), where TP (true positive) is the
number of correctly predicted samples that belong to
the class, TN (true negative) is the number of
correctly predicted samples that do not belong to the
class, FP (false positive) is the number of wrongly
predicted samples that belong to the class and FN
(false negative) is the number of wrongly predicted
samples that do not belong to the class.
Accuracy (CA) calculates the proximity of
measurement results to the true value and gives the
global efficacy of a classifier.
CA = (TP+TN) / (P+N) (1)
Precision (Precision) specifies the positive labels
given by the classifier that are correct.
Precision = TP / (TP+FP) (2)
Recall or sensitivity shows the efficacy of a
classifier to identify positive labels.
Recall = TP/P = TP / (TP+FN). (3)
F1-score (F1) is the harmonic mean of precision
and recall and is between 0 and 1, being 1 the best
value.
F1 = 2 x (Precision x Recall) /
(Precision + Recall)
(4)
Area under the ROC curve (AUC) is the
classifier’s capacity to avoid false classification.
AUC = 1/2 ((TP / (TP+FN))
+ (TN / (FP + TN))
(5)
Three strategies were used, the first one with
classification results obtained by 5 fold cross-
validation and the others two using a separate test
data, one from the same type of cancer and the other
from a different type of cancer.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
210
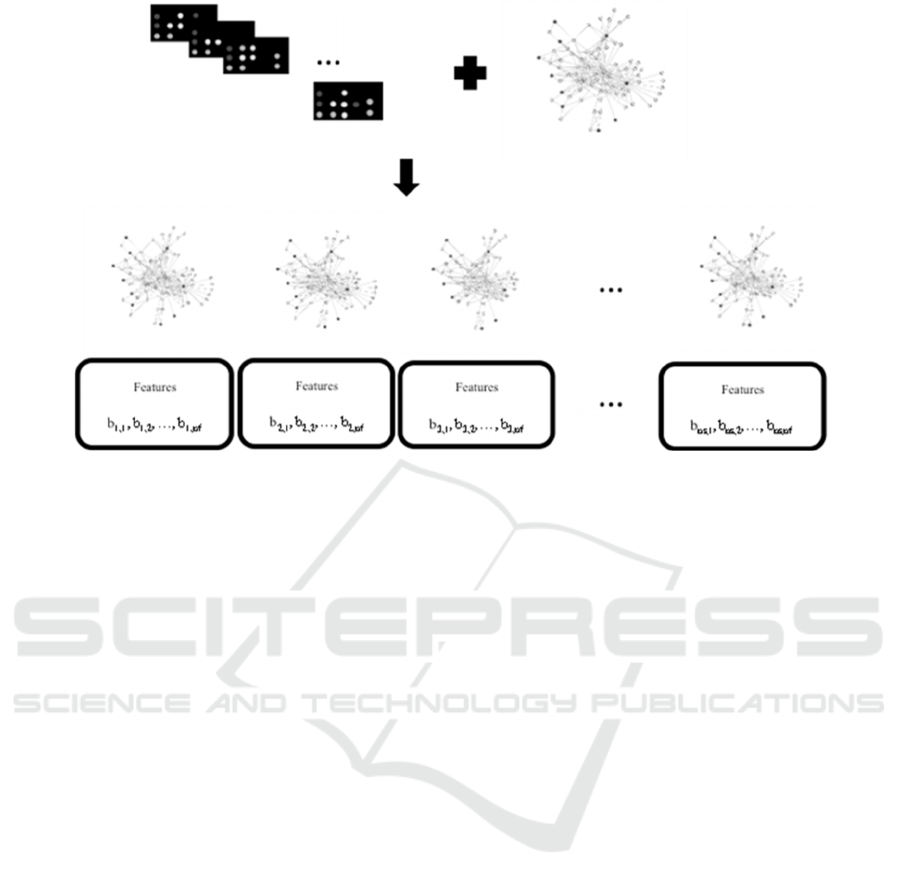
Figure 1: SSN network-based features, where ns is the number of samples and nf is the number of features.
3.4 Network-based Approach
An undirected and unweighted graph G can be
defined as a pair G = (V, E) where V is a set of
vertices representing the nodes and E is a set of edges
representing the connections between the nodes i and
j. The number of nodes of a graph G is denoted by N
and the number of edges of a graph is denoted by L.
Given a graph G = (V, E) the adjacency matrix
representation consists of an N x N matrix A = [a
ij
],
such that a
ij
= 1 if (i, j) belongs to E or a
ij
= 0
otherwise. For undirected graphs the matrix is
symmetric.
Descriptors obtained from the analysis of the SSN
topologies were calculated using the package
NetworkX from Python (Schult and Swart, 2008), the
R package QuACN (Mueller et al., 2011) and the
gtrieScanner (Ribeiro and Silva, 2014) software.
The following methodology was used to obtain
SSN network-based features (Figure 1):
Step 1: Obtain the e matrixes, for e=1,…, ne,
where ne is the number of microarray experiments,
ns
e
is the number of samples of the experiment e and
ng is the number of genes.
EXP
e
= [exp
i
j
], i=1,…, ns
e
; j=1, …, ng (6)
Step 2: Obtain the lists of the top genes ranked by
decreasing order of ReliefF
LTGR
e
, for e=1,..., ne (7)
Step 3: Obtain the union of the previous lists, for
a threshold value, thr_rf that defines the number of
top elements of the lists to be considered.
LUNION = UNION (LTGRe), for
e=1,...,ne
(8)
Step 4: Obtain the submatrices of EXP
e
, for e=1,
..., ne, obtained in step 1., for the genes selected in
step 3.
SEXP
= [sexp
ij
], for i=1,..., sum
e
(ns
e
);
j=1,…, thr_rf
(9)
Step 5: Obtain the lists of the top thr_me most
expressed genes in SEXP, for each sample from e
experiments, for e=1,…, ne.
LGME= [lgme
ij
], for i=1,..., sum
e
(ns
e
);
j=1, .., thr_me
(10)
Step 6: Obtain the lists of the proteins encoded by
the genes of the LGME matrix, P(LGME) for each
sample of the experiments e, for e=1, ..., ne.
LPME= P(LGME)
i
, for i=1,..., sum
e
(ns
e
) (11)
Step 7: Obtain the SSN, the PPI human interaction
sub-networks induced by LPME.
SSN= [ssn
i
], for i=1,..., sum
e
(ns
e
); e=1,…,
ne
(12)
Prediction of Cancer using Network Topological Features
211

Table 1: Group D0 of topological network-based descriptors.
D0.1: Number of nodes D0.9: Size of the largest clique
D0.2: Number of edges D0.10: Number of maximal cliques
D0.3: Density D0.11: Degree assortativity coefficient
D0.4: Number of connected components D0.12: Estrada index
D0.5: Number of nodes of the largest component D0.13: Graph transitivity
D0.6: Number of edges of the largest component D0.14: Average clustering coefficient
D0.7: Diameter of the largest component D0.15: Average shortest path length
D0.8: Global clustering coefficient
Table 2: Groups D1, D2 and D3 of topological network-based descriptors.
D1.1: Wiener D2.1: Total adjacency D3.1: Medium articulation
D1.2: Harary D2.2: Zagreb 1 D3.2: Efficiency
D1.3: BalabanJ D2.3: Zagreb 2 D3.3: Graph index complexity
D1.4: Mean distance deviation D2.4: Modified Zagreb D3.4: Off diagonal
D1.5: Compactness D2.5: Augmented Zagreb D3.5: Spanning tree sensitivity STS
D1.6: Product of row sums D2.6: Variable Zagreb D3.6: Spanning tree sensitivity STSD
D1.7: Hyper distance path index D2.7: Randic
D1.8: Dobrynin eccentricity graph D2.8: Complexity index B
D1.9: Dobrynin avgecc of G D2.9: Normalized edge complexity
D1.10: Dobrynin eccentric graph D2.10: Atom bond connectivity
D1.11: Dobrynin graph integration D2.11: Geometric arithmetic 1
D1.12: Dobrynin unipolarity D2.12: Geometric arithmetic 2
D1.13: Dobrynin variation D2.13: Geometric arithmetic 3
D1.14: Dobrynin centralization D2.14: Narumi Katayama
D1.15: Dobrynin average distance
D1.16: Dobrynin mean distance
vertex deviation
Step 8: Calculate the five groups of topological
properties for each ssn belonging to SSN, where the
number of features, nf, is the number of descriptors
used.
FEAT= [feat
ij
], for i=1,…, sum
e
(ns
e
);
j=1,..., nf; e=1,..., ne
(13)
Several topological measures were included to
capture the structural complexity of the biological
networks. Hereafter are referred the five groups of
descriptors used.
A first group of 15 descriptors, named D0,
calculated using NetworkX (
Table 1). A second group
of 16 descriptors, named D1 that uses distances
between nodes to capture the structural complexity of
the network, a third group, named D2, of 14
descriptors and a forth group of 6 more recent
descriptors, named D3, all of them calculated using
QuACN (Table 2). A fifth group , named D4, of 58
descriptors, where the first 29 are corresponding to
the relative frequency values of 3 to 5 nodes
subgraphs if they are a motif and zero if they are not
a motif and the last 29 are the correspondent z-score
values, which were calculated using 1000 random
networks (
Table 3).
These descriptors were calculated using the
gtriesScanner software. A motif is a subgraph that is
frequent compared to their frequency in a set of
similar random networks. In this paper a subgraph is
considered a motif (Milo et al., 2002), if the
frequency of the subgraph in the network is superior
to 4, the difference between the frequency in the
network (f) and the average frequency in 1000 similar
random networks (avgfr) is greater or equal to 0.10 of
the average frequency in those random networks, and
|z-score| > 2, where z-score = (f - avgfr) / sd, with sd
as the standard deviation.
Table 3: Group D4 of topological network-based
descriptors.
D4.1_j: 3_j_fr
j=1,… 2
D4.4_j: 3_j_zsc
j=1,… 2
D4.2_j: 4_j_fr
j=1,… 6
D4.5_j: 4_j_zsc
j=1,… 6
D4.3_j: 5_j_fr
j=1,… 21
D4.6_j: 5_j_zsc
j=1,… 21
To build the binary classification models three
different supervised learning algorithms were used,
namely the KNN, SVM with RBF kernel and RF
classifiers, all with default parameters. All of the
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
212
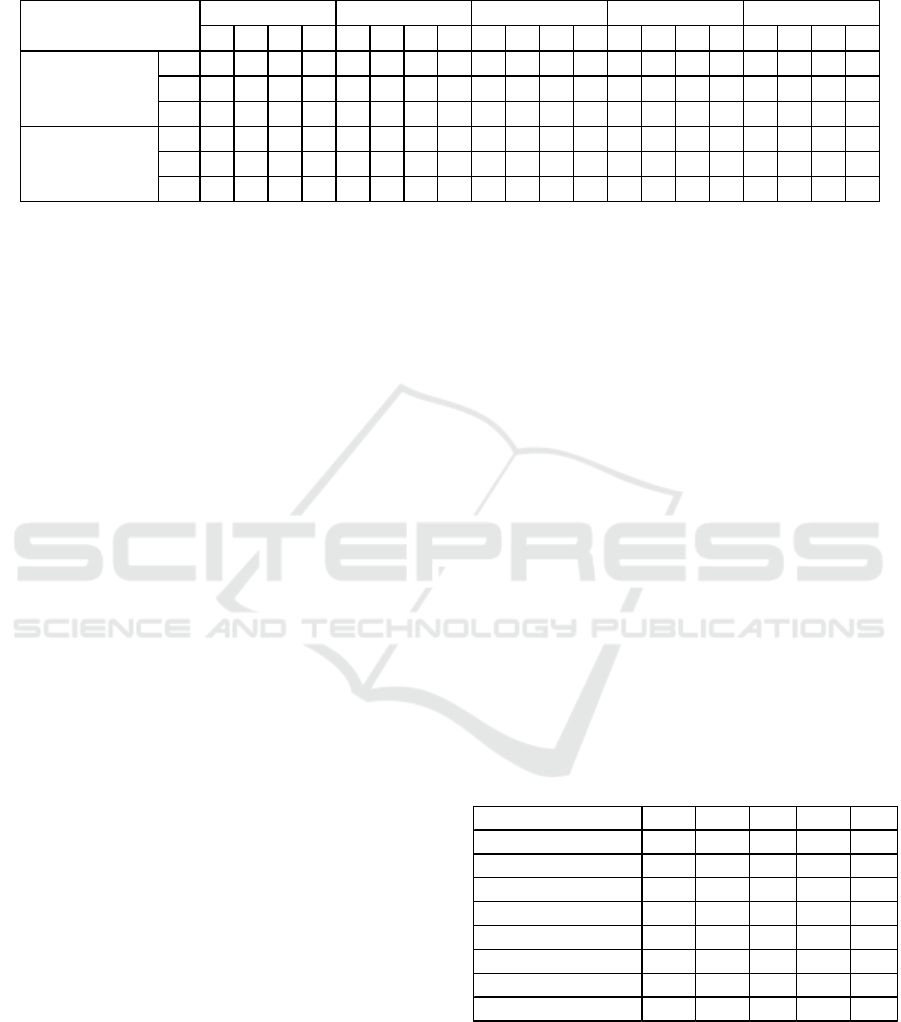
Table 4: Statistical evaluation (CA, Precision, Recall, F1 and AUC) of the binary classification C - Cancer and H- Healthy
for the cases C1, C2, C3 and C4 using the three classifiers KNN, SVM and RF, for all of the network-based features and for
the group of network-based features D4 for the class C.
classifiers used gave similar results, with a slight
advantage in some statistical measures for RF when
using information from breast cancer datasets.
The statistical measures used to evaluate the
performance of the binary classification models were
the CA, the Precision, the Recall, the F1 and the AUC
and values were obtained using three strategies, with
different sets of features groups.
The first strategy, named Case 1 (C1), used 5 fold
cross-validation on the network-based features values
calculated from the three breast cancer microarray
datasets.
The second strategy included two types of tests
that were named Case 2 (C2) and Case 3 (C3) and
here two of the breast cancer datasets were used to
calculate network-based features values for the
training dataset and the remaining one was used to
calculate the network-based features values for the
test dataset. In C2, the training set used E_GEOD-
65194 and E-GEOD-54002 microarray datasets and
the test set used E_GEOD-29044 dataset and in C3,
the training set used E_GEOD-54002 and E-GEOD-
29044 datasets and the test set used E_GEOD-65194
microarray dataset.
The third strategy, named Case 4 (C4), used data
from the three breast cancer microarray datasets for
the training dataset and the liver microarray dataset
was used for the test dataset.
The two sets of features, whose results are shown
in Table 4, are the set of all of the network-based
features (groups D0 to D4) and the set of network-
based features of the group D4. In the case C1, 5-fold
cross validation was used, with results, above 0.95,
for all the statistical measures considered. To test if
the classification obtained in C1 was suffering from
over fitting, different datasets were used as a train set
and as test set, the cases C2 and C3. The results
obtained were, for example for CA, above 0.80 for C2
and above 0.92 for C3, which evidence good
performance of the classifier. The difference between
the values of C2 and C3 may be explained by the
imbalance between cancer and non-cancer samples.
To check if the classification models with datasets
of one type of cancer can be generalized for another
cancer type, the classification model was trained with
data from three breast cancer datasets and tested with
data obtained from a liver cancer dataset, in case C4.
Values obtained and shown in Table 4 are still
positive, probably due to the fact of all being cancer
diseases, but worse than the previous ones.
To analyse which of the network-based features
contributed more for the classification model a
ranking list of features was done. Table 5 shows the
top 5 ranking of the network-based features.
From the analysis of which features are more
informative, it can be stated that the most relevant
features belong mainly to group D0 and group D4.
When all groups of topological features are used as
features variables, it can be seen that the size of the
largest clique and the number of nodes are better
ranked. Motifs of size 4 and 5 are the most
informative motifs.
Table 5: Top five ranking of network-based features.
4 CONCLUSIONS
The statistical evaluation results were obtained using
only topological properties as features variables,
C 1C 2C 3C 4C 1C 2C 3C 4C 1C 2C 3C 4C 1C 2C 3C 4C 1C 2C 3C 4
KNN 0.98 0.80 0.97 0.61 0.98 0.76 0.98 0.64 0.97 0.97 0.99 0.88 0.98 0.85 0.99 0.75 0.96 0.86 0.94 0.55
SVM 0.96 0.81 0.96 0.62 0.99 0.78 0.99 0.70 0.97 0.95 0.96 0.72 0.98 0.86 0.98 0.71 0.98 0.94 0.98 0.64
RF 0.96 0.88 0.98 0.60 0.98 0.88 0.98 0.69 0.98 0.93 1.00 0.69 0.98 0.90 0.99 0.69 0.98 0.94 0.99 0.60
KNN 0.95 0.76 0.92 0.58 0.98 0.73 0.95 0.67 0.97 0.95 0.97 0.68 0.97 0.83 0.96 0.67 0.96 0.82 0.93 0.56
SVM 0.96 0.81 0.96 0.62 0.98 0.79 0.97 0.76 0.97 0.93 0.99 0.60 0.98 0.85 0.98 0.67 0.97 0.87 0.99 0.62
RF 0.96 0.85 0.98 0.57 0.97 0.84 0.98 0.64 0.98 0.93 1.00 0.79 0.98 0.89 0.99 0.71 0.97 0.93 0.99 0.51
D0+D1+D2+ D3+D4
D4
F1 AUC
Cancer
CA Precision Recall
Tests 1st 2nd 3rd 4th 5th
D0+D1+D2+D3+D4 (case 1) D0.9 D0.5 D0.1 D3.2 D4.3_1
D0+D1+D2+D3+D4 (case 2) D4.2_6 D0.1 D0.5 D0.6 D0.2
D0+D1+D2+D3+D4 (case 3) D0.9 D0.4 D4.2_2 D0.13 D0.1
D0+D1+D2+D3+D4 (case 4) D0.9 D0.5 D0.1 D3.2 D4.3_1
D4 (case 1) D4.3_1 D4.1_1 D4.1_6 D4.2_6 D4.5_3
D4 (case 2) D4.2_6 D4.3_17 D4.3_4 D4.3_2 D4.3_1
D4 (case 3) D4.3_18 D4.3_19 D4.2_5 D4.3_17 D4.2_2
D4 (case 4) D4.3_1 D4.1_1 D4.2_1 D4.2_6 D4.5_3
Prediction of Cancer using Network Topological Features
213

measured in the SSN, which are PPI networks built
from the expressed genes without any other biological
information. The results seem to indicate that there
are signatures embedded in the topology dynamics,
modelled through the SSN, which can distinguish
cancer from non-cancer cells for each type of cancer.
This new methodology of creating SSN allows the
capture of the topology dynamics of the system
through the set of samples and allows data to be
reduced and be computationally manageable, keeping
the more informative data, which is supported by the
good results obtained. We consider that this novel
approach is worth and gives different contributions
compared to previous works, namely: the number of
considered topological properties is much higher; the
exclusive use of topological properties (global and
local) with good binary classification results
obtained; the topological dynamics of the system
captured through each sample, different from other
works that use time or states for example, which can
contribute to the capture of different signatures.
The results obtained show that classification
models should be different according to the cancer
disease type considered. More, the knowledge of
which features are more informative can be used, in
the future, to look for signatures based in these
features that could help in the identification of certain
cancer types. Two of the most discriminative features
obtained were the size of the largest clique and motifs
of size 4 and 5. Cliques being fully connected
subnetworks where genes are functionally related and
highly expressed were considered by some
researchers as gene signatures (Pradhan et al., 2012).
The relative frequency and z-score of some motifs as
local topological properties measures, showed to be
discriminatory features, indicating that there are clues
that some small subnetworks could help to distinguish
cancer samples. Adding more biological information
to the more discriminative features found in the
classification, may reveal important signatures like
subgraphs markers of cancer diseases. This approach
also seems worth to be further explored.
Finally the proposed methodology for creating
SSN is a novel contribution that can be extended to
other types of networks, besides PPIs, adding
information that can differentiate samples and capture
their topological dynamics helping to uncover new
signatures that can be biologically relevant for the
identification of diseases.
ACKNOWLEDGEMENTS
This work has received support from the RD-
CONNECT European project (EC contract number
305444).
REFERENCES
Amberger, J. S., Bocchini, C. A., Schiettecatte, F., Scott, A.
F. & Hamosh, A., 2015. Omim. Org: Online Mendelian
Inheritance In Man (Omim®), An Online Catalog Of
Human Genes And Genetic Disorders. Nucleic Acids
Research, 43, D789-D798.
Arrais, J. P. & Oliveira, J. L., 2011. Using Biomedical
Networks To Prioritize Gene-Disease Associations.
Open Access Bioinformatics, 1, 123-130.
Ay, A., Gong, D. & Kahveci, T., 2014. Network-Based
Prediction Of Cancer Under Genetic Storm. Cancer
Informatics, 13, 15.
Barabási, A.-L., Gulbahce, N. & Loscalzo, J., 2011.
Network Medicine: A Network-Based Approach To
Human Disease. Nature Reviews. Genetics, 12, 56-68.
Barabasi, A.-L. & Oltvai, Z. N., 2004. Network Biology:
Understanding The Cell's Functional Organization.
Nature Reviews Genetics, 5, 101-113.
Barter, R., Schramm, S.-J., Mann, G. & Yang, Y. H., 2014.
Network-Based Biomarkers Enhance Classical
Approaches To Prognostic Gene Expression
Signatures. Bmc Systems Biology, 8, S5.
Chen, D. & Yang, H., 2014. Comparison Of Gene
Regulatory Networks Of Benign And Malignant Breast
Cancer Samples With Normal Samples. Genetics And
Molecular Research: Gmr, 13, 9453.
Chuang, H. Y., Lee, E., Liu, Y. T., Lee, D. & Ideker, T.,
2007. NetworkBased Classification Of Breast Cancer
Metastasis. Molecular Systems Biology, 3, 140.
Consortium, T. U., 2014. Activities At The Universal
Protein Resource (Uniprot). Nucleic Acids Research,
42, D191-D198.
Demsar, J., Zupan, B. & Leban, G., 2007. Orange: From
Experimental Machine Learning To Interactive Data
Mining. White Paper, Faculty Of Computer And
Information Science, University Of Ljubljana (2004).
Dennis, G., Jr., Sherman, B. T., Hosack, D. A., Yang, J.,
Gao, W., Lane, H. C. & Lempicki, R. A., 2003. David:
Database For Annotation, Visualization, And
Integrated Discovery. Genome Biol, 4, P3.
Dezfuly, M. & Sajedi, H,. 2015. Predict Survival Of
Patients With Lung Cancer Using An Ensemble Feature
Selection Algotithm And Classification Methods In
Data Mining. Journal Of Information, 1, 1-11.
Dominietto, M., Tsinoremas, N. & Capobianco, E., 2015.
Integrative Analysis Of Cancer Imaging Readouts By
Networks. Molecular Oncology, 9, 1-16.
Farkas, I. J., Korcsmáros, T., Kovács, I. A., Mihalik, Á.,
Palotai, R., Simkó, G. I., Szalay, K. Z., Szalay-Beko,
M., Vellai, T. & Wang, S., 2011. Network-Based Tools
For The Identification Of Novel Drug Targets. Sci
Signal, 4, Pt3.
Furey, T. S., Cristianini, N., Duffy, N., Bednarski, D. W.,
Schummer, M. & Haussler, D. 2000. Support Vector
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
214

Machine Classification And Validation Of Cancer
Tissue Samples Using Microarray Expression Data.
Bioinformatics, 16, 906-914.
Jonsson, P. F. & Bates, P. A., 2006. Global Topological
Features Of Cancer Proteins In The Human
Interactome. Bioinformatics, 22, 2291-7.
Kononenko, I. Estimating Attributes: Analysis And
Extensions Of Relief. Machine Learning: Ecml-94,
1994. Springer, 171-182.
Menche, J., Sharma, A., Kitsak, M., Ghiassian, S. D., Vidal,
M., Loscalzo, J. & Barabási, A.-L., 2015. Uncovering
Disease-Disease Relationships Through The
Incomplete Interactome. Science, 347, 1257601.
Milo, R., Shen-Orr, S., Itzkovitz, S., Kashtan, N.,
Chklovskii, D. & Alon, U., 2002. Network Motifs:
Simple Building Blocks Of Complex Networks. Science,
298, 824-827.
Mueller, L., Kugler, K., Graber, A., Emmert-Streib, F. &
Dehmer, M., 2011. Structural Measures For Network
Biology Using Quacn. Bmc Bioinformatics, 12, 492.
Nancy, S. G. & Alias Balamurugan, S. A., 2013. A
Comparative Study Of Feature Selection Methods For
Cancer Classification Using Gene Expression Dataset.
Journal Of Computer Applications (Jca), 6, 2013.
Oti, M., Snel, B., Huynen, M. A. & Brunner, H. G., 2006.
Predicting Disease Genes Using Protein–Protein
Interactions. Journal Of Medical Genetics, 43, 691-
698.
Ou-Yang, L., Dai, D.-Q., Li, X.-L., Wu, M., Zhang, X.-F.
& Yang, P., 2014. Detecting Temporal Protein
Complexes From Dynamic Protein-Protein Interaction
Networks. Bmc Bioinformatics, 15, 335.
Pradhan, M. P., Nagulapalli, K. & Palakal, M. J., 2012.
Cliques For The Identification Of Gene Signatures For
Colorectal Cancer Across Population. Bmc Systems
Biology, 6, S17.
Pržulj, N., 2007. Biological Network Comparison Using
Graphlet Degree Distribution. Bioinformatics, 23,
E177-E183.
Pržulj, N., Corneil, D. G. & Jurisica, I., 2004. Modeling
Interactome: Scale-Free Or Geometric?
Bioinformatics, 20, 3508-3515.
Ramani, R. G. & Jacob, S. G., 2013. Improved
Classification Of Lung Cancer Tumors Based On
Structural And Physicochemical Properties Of Proteins
Using Data Mining Models. Plos One, 8, E58772.
Ribeiro, P. & Silva, F,. 2014. G-Tries: A Data Structure For
Storing And Finding Subgraphs. Data Mining And
Knowledge Discovery, 28, 337-377.
Robnik-Šikonja, M. & Kononenko, I., 2003. Theoretical
And Empirical Analysis Of Relieff And Rrelieff.
Machine Learning, 53, 23-69.
Saeys, Y., Inza, I. & Larrañaga, P., 2007. A Review Of
Feature Selection Techniques In Bioinformatics.
Bioinformatics, 23, 2507-2517.
Schult, D. A. & Swart, P. Exploring Network Structure,
Dynamics, And Function Using Networkx. Proceedings
Of The 7th Python In Science Conferences (Scipy
2008), 2008. 11-16.
Siegel, R. L., Miller, K. D. & Jemal, A., 2015. Cancer
Statistics, 2015. Ca: A Cancer Journal For Clinicians,
65, 5-29.
Sokolova, M. & Lapalme, G., 2009. A Systematic Analysis
Of Performance Measures For Classification Tasks.
Information Processing & Management, 45, 427-437.
Sonachalam, M., Shen, J., Huang, H. & Wu, X., 2012.
Systems Biology Approach To Identify Gene Network
Signatures For Colorectal Cancer. Frontiers In
Genetics, 3.
Trapé, A. P. & Gonzalez-Angulo, A. M., 2012. Breast
Cancer And Metastasis: On The Way Toward
Individualized Therapy. Cancer Genomics -
Proteomics, 9, 297-310.
Vidal, M., Cusick, M. E. & Barabasi, A.-L., 2011.
Interactome Networks And Human Disease. Cell, 144,
986-998.
Wang, J., Zuo, Y., Man, Y.-G., Avital, I., Stojadinovic, A.,
Liu, M., Yang, X., Varghese, R. S., Tadesse, M. G. &
Ressom, H. W., 2015. Pathway And Network
Approaches For Identification Of Cancer Signature
Markers From Omics Data. Journal Of Cancer, 6, 54.
Yu, H., Lin, C.-C., Li, Y.-Y. & Zhao, Z., 2013. Dynamic
Protein Interaction Modules In Human Hepatocellular
Carcinoma Progression. Bmc Systems Biology, 7, S2.
Prediction of Cancer using Network Topological Features
215
